- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Structural biology
- Targeting Acute Pancreatitis with inhibitors of kynurenine-3-monooxygenase that tilt the catalytic flavin
Targeting Acute Pancreatitis with inhibitors of kynurenine-3-monooxygenase that tilt the catalytic flavin
X-ray crystallographic and mechanistic studies have enabled the discovery of kynurenine-3-monooxygenase (KMO) inhibitors suitable for preclinical evaluation in Acute Pancreatitis. These picomolar, long-residence-time compounds trap the catalytic flavin in an unusual tilted orientation that overcomes the mechanistic liability of stimulated hydrogen peroxide production seen with previous active site binders.
Scientists at GlaxoSmithKline (GSK) and the University of Edinburgh have worked closely together to find new inhibitors of kynurenine-3-monooxygenase (KMO) to treat Acute Pancreatitis.
Acute Pancreatitis (AP) disease is typically caused by gallstones or excessive alcohol consumption. For the majority of AP patients, symptoms resolve within a week or so. However, in approximately one in five cases, the disease progresses to multiple organ failure with a > 20% mortality rate. Currently there are no disease-modifying treatments available, highlighting the need for new therapies.
In mammals, the major metabolic route of tryptophan degradation is via the kynurenine pathway. KMO is a class A flavoprotein monooxygenase (FPMO) that catalyses the hydroxylation of L-kynurenine (L-Kyn) using NADPH as a co-substrate. The product, 3-hydroxy kynurenine (3-HK), is a neurotoxin that can also induce apoptotic cell death in endothelial cells. Pharmacological inhibition of KMO has long been considered as a target for neurogenerative disorders such as Huntington’s and Alzheimer’s diseases, as well as more recently, for Acute Pancreatitis Multiple Organ Dysfunction Syndrome (AP-MODS).
Surprisingly for such an important metabolic enzyme and drug target, structural insights have so far been remarkably limited. Human KMO is a 56 kDa protein containing a tightly bound FAD co-factor and is attached to the mitochondrial outer membrane via a C-terminal region. Human KMO remains recalcitrant to crystallisation with no structure reported to date. The homologous bacterial Pseudomonas fluorescens enzyme, Pf-KMO, (34% sequence identity), which lacks the transmembrane region, was therefore developed as a structural surrogate.
Native Pf-KMO crystals diffracted poorly, but a double C252S/C461S Pf-KMO mutant could be optimised to yield a high resolution-diffracting monoclinic crystal form amenable for ligand soaking studies, enabling a rapid turnaround of crystal structures determined on ID23-1, ID30A and ID30B to drive structure-based drug design.
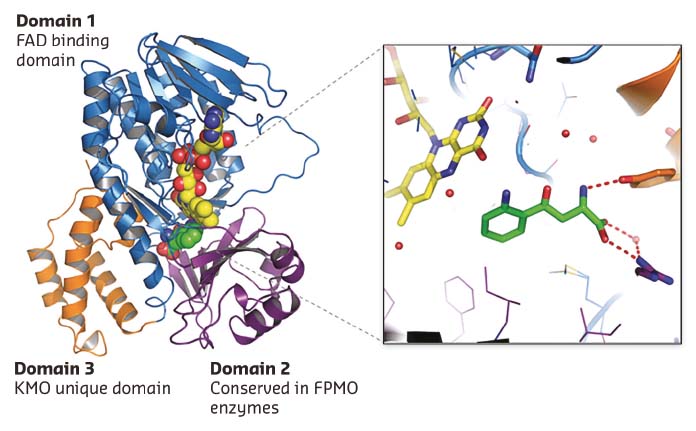
Many KMO inhibitors are substrate inspired. A 1.5 Å L-Kyn bound Pf-KMO structure gave the first view of how the native substrate sits within the active site (Figure 29). The details of this structure differ from previous computational models and represent a significant advance, allowing explanation of puzzling structure-activity relationships and providing a firm basis for novel inhibitor discovery and mechanistic analysis. This was demonstrated by the discovery of the first type-II KMO inhibitors that bind to an unprecedented tilting flavin conformation of Pf-KMO.
 |
|
Fig. 29: 1.5 Å X-ray structure of Pf-KMO in complex with substrate. |
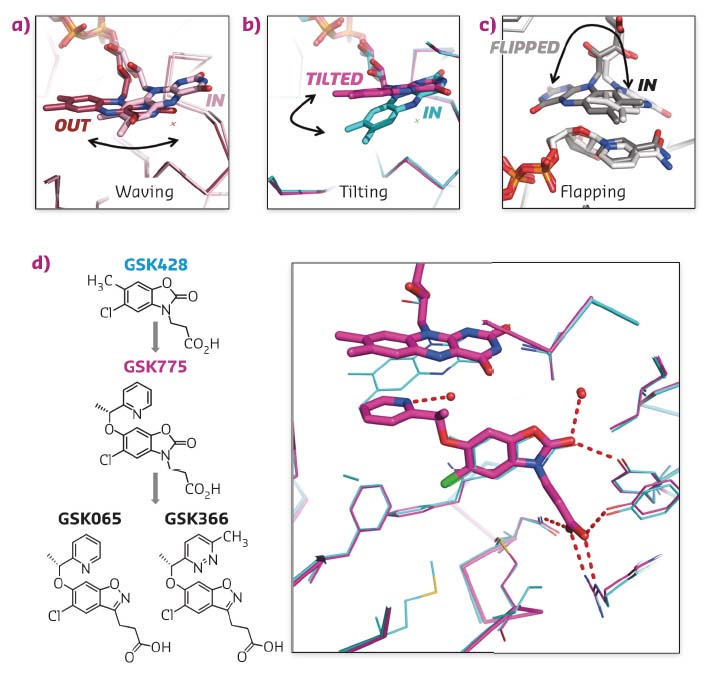
Carefully choreographed flavin motions are critical to multiple steps in the catalytic cycle of FPMO enzymes, controlling substrate binding, product release and regulating the reductive and oxidative steps during catalysis. Rare snapshots of flavin motion have been captured by crystallography (Figures 30a, c) but none of these were known to be important or benefical for inhibitor design.
 |
|
Fig. 30: a-c) Crystal structures of flavin captured in different orientations in various crystal structures. d) Chemical structures of type I inhibitor GSK428 and type II inhibitors GSK775, GSK065 and GSK366. Overlay of X-ray structures of Pf-KMO bound with GSK428 (cyan) and GSK775 (magenta). |
GSK775 was synthesised during a structure-based optimisation of a series of oxazolidinone inhibitors such as GSK428 (Figure 30d). The pyridine ring of GSK775 was designed to occupy space close to the flavin ring to gain additional potency. However, GSK775 was much more potent than expected and exhibited slow kinetics. Crystal structures revealed it didn’t bind close to the flavin but, instead, pushed it into a tilted orientation never seen before (Figures 30b, d). This traps the flavin in an unproductive position that decouples substrate site binding from NAPDH reduction and flavin oxidation. The result is that type-II compounds such as GSK775 lose the mechanistic liability of stimulated hydrogen peroxide production carried by simple substrate site inhibitors such as GSK428.
Transferring this structural and mechanistic understanding to a benzisoxazole series has delivered picomolar, long residence time, type-II compounds, GSK065 and GSK366, which have the full profile of pharmaceutical and pharmacological in vitro and in vivo properties for progression to clinical evaluation in AP-MODS.
Principal publication and authors
Structural and mechanistic basis of differentiated inhibitors of the acute pancreatitis target kynurenine-3-monooxygenase, J.P. Hutchinson (a), P. Rowland (a), M. Taylor (b), E.M. Christodoulou (a), C. Haslam (a), C.M. Hobbs (a), D.S. Holmes (c), P. Homes (a), J. Liddle (c), D.J. Mole (d, e), I. Uings (c), A.L. Walker (c), S.P. Webster (f), C. G. Mowat (b) and C. Chung (a), Nat Commun 8, 15827 (2017); doi:10.1038/ncomms15827.
(a) Platform Technologies and Science , GlaxoSmithKline, Stevenage (UK)
(b) EastChem School of Chemistry, University of Edinburgh (UK)
(c) Discovery Partnerships with Academia, GlaxoSmithKline, Stevenage (UK)
(d) Medical Research Council Centre for Inflammation Research, Edinburgh (UK)
(e) Clinical Surgery, University of Edinburgh (UK)
(f) British Heart Foundation Centre for Cardiovascular Science, University of Edinburgh (UK)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.