- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Structural biology
- Structure-based development of small molecules that disrupt glycosomal transport in Trypanosoma parasites
Structure-based development of small molecules that disrupt glycosomal transport in Trypanosoma parasites
PEX14 and PEX5 are crucial to the transport of metabolic enzymes into glycosomes in Trypanosoma protists. Inhibition of the complex formed between these two proteins disrupts essential metabolic processes and leads to parasite death. Using a structure-based approach, the first PEX14-PEX5 protein-protein interaction inhibitors have been developed. These disrupt glycosomal transport and kill Trypanosoma.
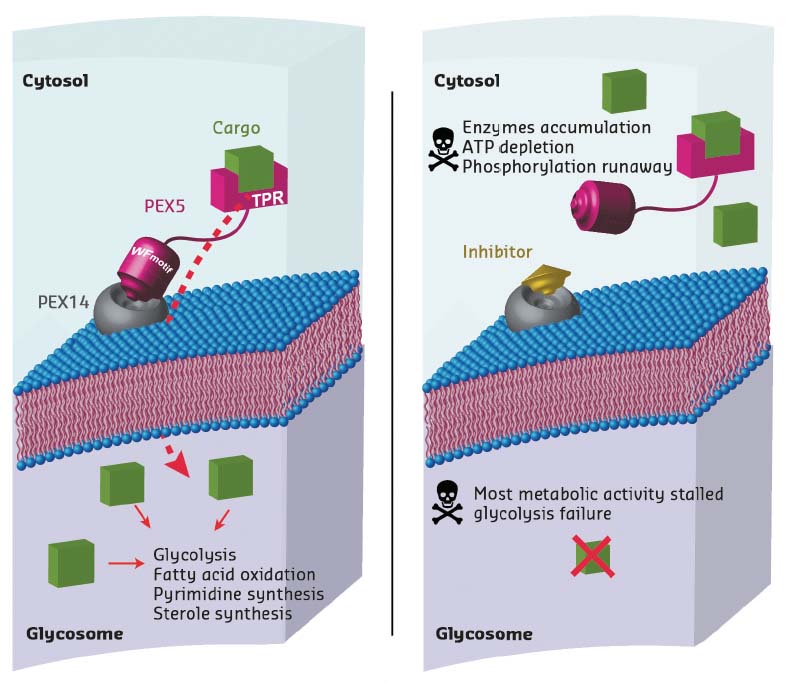
Trypanosoma parasites are protozoa that cause numerous diseases including sleeping sickness and Chagas disease. Unlike for other eukaryotic cells, glycolytic processes in Trypanosoma parasites take place within a single organelle, the glycosome. As this organelle lacks genetic information, all the metabolic enzymes need to be imported post-translationally. The PEX14-PEX5 protein-protein interaction plays a crucial role in this pathway. Consequently, inhibiting the formation of the complex between these two proteins is a way of disrupting glycosome function, leading to accumulation of glycosomal enzymes in the cytosol which, in turn, causes adenosine triphosphate (ATP) depletion and metabolic collapse, resulting in Trypanosoma parasite death (Figure 8).
 |
|
Fig. 8: Principle of the mechanism of action of PEX14 inhibitors, showing a normal cell (left) and a cell in the presence of an inhibitor (right). The inhibitor molecule interferes with PEX5 binding to the membrane-associated import factor PEX14. Instead of being transported to the glycosome, enzymes are stranded in the cytosol, multiplying damage to the cell. This disrupts major metabolic pathways in Trypanosoma, causing ATP depletion and, eventually, cell death. |
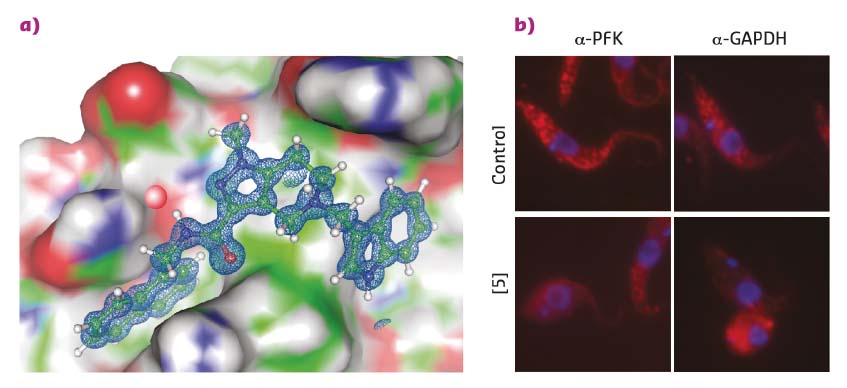
Existing structural information [1] was employed to perform a 3D pharmacophore-based in silico screening of a virtual compound library followed by 3D docking. This identified drug-like pyrazolo[4,3-c]pyridine derivatives that bind to PEX14 and disrupt the PEX14-PEX5 interface. Hit validation by NMR binding assays and hit optimisation based on results from NMR fragment screening led to more potent molecules that were amenable to cocrystalisation with the PEX14 protein. High-resolution X-ray data were collected at beamlines ID29 and ID30b with crystals of the resulting PEX14-inhibitor complexes yielding exceptional data quality to a resolution of up to 0.86 Å (Figure 9a). These high-resolution structures unambiguously confirmed the in silico derived binding model of the inhibitors and enabled further rational compound optimisation.
 |
|
Fig. 9: a) A PEX14 inhibitor bound to trypanosomal PEX14. Very high-resolution structural data allowed the rapid optimisation of inhibitors towards enhanced potency and selectivity. b) Fluorescence microscopy reveals the disruption of the glycosomal transport system by PEX14 inhibitors. Enzymes that are localised in glycosomes in control cells (granular pattern) are spread over the entire cell volume in the presence of the inhibitor. |
A peculiarity of the cocrystal structures is the presence of a structural water molecule that mediates the interactions of ligands with the PEX14 protein and was not predicted by docking. The importance of this water molecule was evident as it could not be displaced from the protein surface by polar moieties that were tentatively attached to the ligand. Furthermore, the water molecule interacts with the side chain of Asn31, an amino acid residue that is specific to Trypanosoma and distinct from humans, potentially contributing to ligand specificity. Detailed analysis of the water network surrounding the protein-inhibitor complex was necessary to understand the binding energetics and optimise the ligands.
The PEX14 inhibitors that were developed show potent and selective in vitro activity against clinically relevant T. brucei and T. cruzi species. This activity correlates with the inhibitory effects of the ligands against PEX14-PEX5 complex formation. Together with the observed mislocalisation of glycosomal enzymes to the cytosol and decreased cellular ATP levels, this supports an on-target mode of these inhibitors (Figure 9b).
Many of the glycosomal enzymes have individually been successfully evaluated as trypanocidal agents in the past. This work shows an efficient way to disrupt the function of all of these enzymes simultaneously and validates the PEX14-PEX5 protein-protein interaction interface as a viable drug target for Trypanosoma infections.
Principal publication and authors
Inhibitors of PEX14 disrupt protein import into glycosomes and kill Trypanosoma parasites, M. Dawidowski (a, b), L. Emmanouilidis (a, b), V.C. Kalel (c), K. Tripsianes (d), K. Schorpp (e), K. Hadian (e), M. Kaiser (f, g), P. Mäser (f, g), M. Kolonko (a), S. Tanghe (h), A. Rodriguez (h), W. Schliebs (c), R. Erdmann (c), M. Sattler (a, b) and G.M. Popowicz (a, b), Science 355, 1416-1420 (2017); doi: 10.1126/science.aal1807.
(a) Institute of Structural Biology, Helmholtz Zentrum München, Neuherberg (Germany)
(b) Center for Integrated Protein Science Munich, Chair of Biomolecular NMR, Department Chemie, Technische Universität München, Garching (Germany)
(c) Institute of Biochemistry and Pathobiochemistry, Department of Systems Biochemistry, Ruhr University Bochum (Germany)
(d) CEITEC, Central European Institute of Technology, Masaryk University, Brno (Czech Republic)
(e) Assay Development and Screening Platform, Institute of Molecular Toxicology and Pharmacology, Helmholtz Zentrum München, Neuherberg (Germany)
(f) Swiss Tropical and Public Health Institute, Basel (Switzerland)
(g) University of Basel (Switzerland)
(h) New York University School of Medicine, Department of Microbiology, New York (USA)
References
[1] C. Neufeld et al., EMBO J. 28, 745-754 (2009).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.