- Home
- News
- General News
- Serial crystallography...
Serial crystallography develops by leaps and bounds at the ESRF
06-04-2018
Serial crystallography is a new way of studying macromolecular structures using synchrotron and X-FEL sources around the world. The Structural Biology group at the ESRF is continuously developing new methods to advance the field. Two articles describing advances made are published today in Acta Crystallographica Section D.
“On the Structural Biology Group beamlines one of the ultimate aims is that users can define protocols for experiments, click ‘go’ and let the experiments run by themselves”, explains Gordon Leonard, head of the Structural Biology group at the ESRF. With this idea in mind and to get as much information as possible from the samples available, the team has already adopted serial crystallography, a technique which involves taking diffraction data from many, sometimes hundreds or thousands, of crystals in order to assemble a complete dataset, piece by piece. Indeed, the members of the group are constantly developing new ways to improve the method through collaboration involving scientists from the ESRF, DESY, the Hamburg Centre for Ultrafast Imaging, the European X-FEL and the University of Hamburg.
Turning hindrance to results
In an X-ray crystallography experiment, two critical pieces of information are required in order to determine the atomic structure of a molecule: reflection intensities and reflection phases. The former can be directly measured, while the latter must be obtained indirectly. For example, molecular replacement (using a similar molecule’s structure as a model) or the introduction of heavy atoms that will bind to protein can be used to this end. Determining these phases can be a laborious process if molecular replacement is unsuccessful. A second major problem is that when a molecule is irradiated at a synchrotron source, radiation damage is inevitable. “Some parts of the molecule are more susceptible to radiation, like disulphide bridges or heavy atoms, and they get damaged first”, explains Max Nanao, main author of the publication. “One idea is to use that information on what are the more susceptible parts to determine phases for the reflections”, he adds. This method, called radiation damage-induced phasing (RIP) was first explored at the ESRF in 2002 and has been developed in the ensuing years at the ESRF and other synchrotrons. One major problem with the technique is that researchers straddle a delicate line between obtaining good quality data (by putting more photons on the crystals) and not overly damaging the crystals. The advantage of the serial approach is that by using many crystals, one can significantly improve the quality of the data at a given X-ray dose. However, it was unknown whether the differences between crystals would swamp the phasing signal from radiation damage.
It took Max Nanao, Nicolas Foos and their collaborators two years of work to overcome this issue. They did it by comparing data from 2 exposures of X-rays across many crystals –some highly damaged, others relatively undamaged- and by using the differences to locate sites of radiation damage and then, based on these, determine ph ases.
The results of Nanao and his colleagues mean that maybe in the future there won’t be a need to add heavy atoms to molecules or to work by molecular replacement anymore. “This technique further opens avenues for phasing the crystal structures of native proteins”, says Gordon Leonard.
MESHBEST or picking up the best diffraction patterns
A common serial crystallography technique, developed at ESRF and EMBL Hamburg, is the “mesh and collect” method in which the first step is the harvesting of many small crystals directly from crystallisation drops, using a standard sample holder. The sample holder is then scanned in two dimensions at cryogenic temperatures using a low intensity X-ray mesh scan to automatically determine the positions of crystals by X-ray diffraction. From those crystals, partial diffraction data sets are recorded over a small rotation range. After integration, the individual partial data sets are sorted in clusters of isomorphous similarity, which are then merged to give a complete diffraction data set that can be used for structure solution.
One of the problems with “mesh and collect” is that scientists don’t know a priori how many crystals a diffraction pattern relates to, in other words, if the pattern is a result of crystals placed on top of each other. Samples may vary in size, shape and diffraction strength. Scientist Alexander Popov, together with post-doctoral researcher Igor Melnikov and EMBL Hamburg scientist Gleb Bourenkov, came up with the method and software “MeshBest”. The program automatically analyses diffraction images collected during a mesh scan and produces a two-dimensional crystal map showing estimates of the dimensions, centre positions and diffraction qualities of each crystal contained in the mesh area. Sample regions producing diffraction images resulting from the superposition of more than one crystal are distinguished from regions with single-crystal diffraction. Such information is critical for the proper organisation and design of subsequent diffraction data collection protocols.
The question as to which datasets are best combined to produce a complete dataset is a crucial one. A software called ccCluster, developed by ESRF Junior Scientist Gianluca Santoni, allows researchers to visualise in a straightforward way which partial datasets should be combined. The software will soon be included in the CCP4 programme suite used by many protein crystallographers around the world.
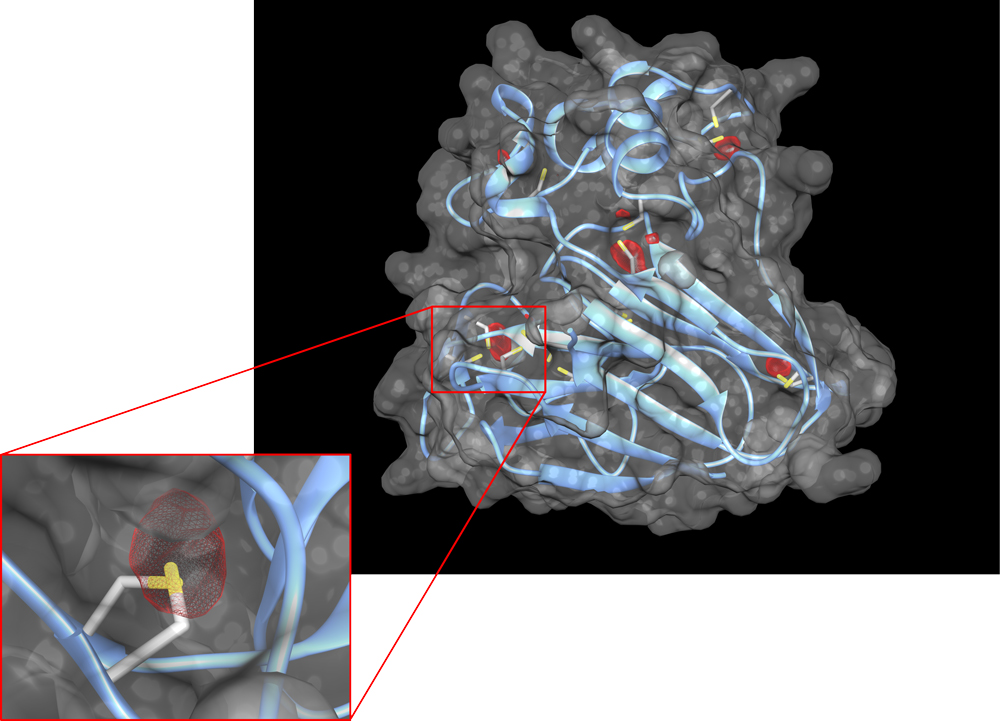 |
|
Thaumatin X-ray structure at 1.4 A resolution. In blue is shown the main chain of thaumatin, side chain atoms are colored by element for Cysteine and Methionine residues. The dark gray envelop represents the protein volume (based on atomic radii). In red meshes are shown the damaged positions. Theses areas of specific radiation damage are used for phase determination. Focus is done on one of the damaged area. Credits: Nicolas Foos. |
Injecting samples into the beam
Today, most samples studied on macromolecular crystallography beamlines at synchrotron sources are frozen at cryogenic temperatures. This slows down the damage resulting from exposure to X-rays but sometimes limits the conformational space a molecule can adopt. “At room temperature the molecules have a higher mobility and they can show alternative conformations”, explains Christoph Mueller-Dieckmann, deputy head of the Structural Biology group. However, the challenges for room temperature analysis are multiple such as dehydration or a greatly enhanced susceptibility to radiation damage. To try to overcome these limitations, and in collaboration with the team of Ilme Schlichting from the MPI of Medical Research in Heidelberg and researchers from the IBS, ILL and EMBL in Grenoble, a grease injector, originally developed for XFEL experiments by the MPI team is used to continuously flow crystals suspended in a high viscosity carrier through the X-ray beam. Initial results are extremely encouraging. However, and as Mueller-Dieckmann also explains, “for our injector experiments to be most efficient, we need small beams with lots of photons, like the ones we will have once the Extremely Brilliant Source (EBS) project is complete”.
 |
|
Daniele de Sanctis on the ID29 beamline. Credits: S. Candé. |
The new EBS beamline
In the coming years, and in parallel with the Extremely Brilliant Source project, a new beamline on Serial Crystallography will be built at the ESRF. The new ID29-SSX (Synchrotron Serial Crystallography) beamline will profit from the ESRF’s Extremely Brilliant Source by delivering an unequalled flux density in a submicron beam size, over a large energy range. It is designed to study micro-crystals that are delivered with different methods (Injectors, Fixed target supports, microfluidic devices are examples) mainly at room temperature. Researchers hope it will exploit time-dependent functional conformation changes upon ligand binding and open new paths in drug discovery research, thanks to rapid ligand diffusion in microcrystals. “The new ID29 is to revolutionize macromolecular crystallography at synchrotrons. We are aiming to an increase of flux density by six orders of magnitude. Such a ‘hot’ beam will make it possible to collect data from yet smaller crystalline samples in a microsecond. With this time resolution we will be able to observe protein at work, as in a molecular movie”, explains Daniele de Sanctis, in charge of the new beamline.
References:
Melnikov, I., et al., (2018). Acta Cryst. D74, https://doi.org/10.1107/S2059798318002735.
Foos, N., et al., (2018). Acta Cryst. D74, https://doi.org/10.1107/S2059798318001535.
Text by Montserrat Capellas Espuny
Top image: A slab through the crystal structure of Thaumatin, showing one of the locations of specific radiation damage that were used for phasing. Credits: M. Nanao.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.