- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Structural biology
- SMART-420 reverses antibiotic resistance in Mycobacterium tuberculosis
SMART-420 reverses antibiotic resistance in Mycobacterium tuberculosis
Resistance to the current generation of antibiotics is an increasing threat to human health and strains of drug-resistant Mycobacterium tuberculosis have spread around the globe. SMARt-420 (Small Molecule Aborting Resistance) fully reverses the ethionamide (ETH)-acquired resistance of M. tuberculosis by activating an alternative ETH bioactivation pathway in the bacterium.
Antibiotic resistance is a serious, global threat to human health. Although cases of tuberculosis (TB) are decreasing by ~1.5% a year, the rise of drug resistant-TB (DR-TB), which already causes around 250,00 deaths per year (2015 figures), may reverse this and cases of TB will start to increase. Many important anti-TB therapeutics are prodrugs. Here, bacterial resistance mechanisms are often the result of mutations in the bacterial enzyme pathway targeted or the acquisition by these pathways of enzymes that modify or degrade the drug molecule introduced [1]. Sometimes, these resistance mechanisms can be reversed by combining the drug with other molecules (i.e. β-lactams plus calvulanic acid [2]). However, examples of this phenomenon are few and far between.
Many of the currently most efficient anti-TB antibiotics such as isoniazid (INH), pyrazinamide (PZA), p-aminosalicylic acid (PAS), ethionamide (ETH) and the recently approved delamanid (OPC-67683) are prodrugs requiring bioactivation by M. tuberculosis. For example, the bioactivation of ETH depends on the Baeyer-Villiger monooxygenase EthA, the production of which is regulated by the TetR-type transcriptional repressor EthR. In ETH-sensitive clinical strains of TB the effect of the prodrug can be increased during treatment by combining it with the small molecule BDM41906, which boosts the production of EthA by inhibiting the regulatory effect of EthR. However, as clinical resistance to ETH occurs via mutations in EthA, which decrease the level of ETH bioactivation, boosting the production of this mutated enzyme does not revert resistance.
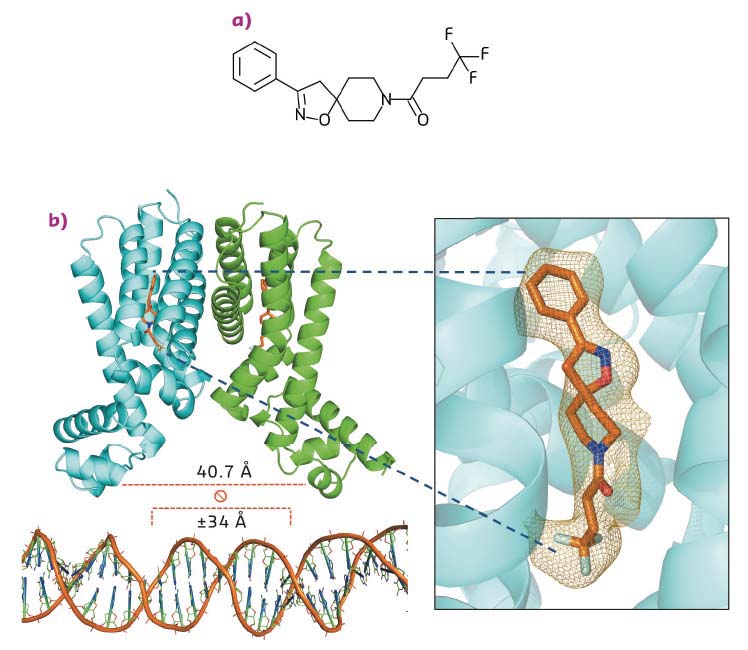
A potential mechanism for reversing or attenuating drug resistance in TB is the induction of alternative bioactivation pathways. In the manuscript on which this article is based, it is shown that the Small Molecules Aborting Resistance (SMARt) family of spiroisoxazolines reverses the resistance of M. tuberculosis to ETH by activating alternative bioactivation pathways regulated by protein EthR2. Thermal shift assays strongly suggested that the SMARt family member SMARt-420 interacts directly with EthR2. The full details of this interaction were revealed by a series of X-ray crystallography experiments carried out in-house, at SOLEIL and at the ESRF (ID23-1). The crystal structures obtained (Figure 21) revealed EthR2 to be an archetypal TetR-type transcription regulator made up of a homodimer containing a pair of helix-turn-helix (HTH) motifs involved in the binding of EthR2 to its target DNA region. In TetR-type regulators, ligands bound to each partner of the homodimer induce a conformational change in the HTH motifs, modifying the binding of the regulator to its target DNA. The crystal structure of the EthR2–SMARt-420 complex revealed that binding of the ligand increased the distance between the HTH motifs to ~ 40.7 Å, far larger than the ±34 Å required for the efficient binding of such regulators to DNA. This observation straightforwardly provides the mechanism of action of SMARt-420.
 |
|
Fig. 21: a) A chemical representation of the spiroisoxazoline SMARt-420, which reverses ETH resistance in M. tuberculosis. b) A ribbon representation of the crystal structure of the EthR2/SMARt-420 complex. The ligands are shown as yellow sticks. The inset shows an omit map, contoured at 1.2 x r.m.s., illustrating the mode of binding of SMARt-420 to an EthR2 monomer. The binding of SMARt-420 to EthR2 increases the distance between the HTH binding motifs to 40.7 Å, sterically inhibiting the binding of EthR2 to its target DNA (bottom). |
The reversal of ETH resistance by SMARt-420 was confirmed in a series of experiments not described here. These suggest that the development of therapeutic protocols based on the switching between ETH and the “ETH + SMARt-420” combination could prove very efficient in the treatment of TB, providing the potential to eliminate populations of ETH-resistant bacteria that may emerge during treatments with this traditional anti-TB antibiotic.
Principal publication and authors
Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420, N. Blondiaux (a), M. Moune (a), M. Desroses (b, c) R. Frita (a), M. Flipo (b), V. Mathys (d), K. Soetaert (d), M. Kiass (d), V. Delorme (a, e), K. Djaout (a), V. Trebosc (f, g), C. Kemmer (f), R. Wintjens (h), A. Wohlkönig (i, j), R. Antoine (a), L. Huot (a), D. Hot (a), M. Coscolla (k, l), J. Feldmann (k, l), S. Gagneux (k, l), C. Locht (a), P. Brodin (a), M. Gitzinger (f), B. Déprez (b), N. Willand (b) and A. Baulard (a), Science 355, 1206-1211 (2017); doi: 10.1126/science.aag1006.
(a) Université Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille (France)
(b) Université Lille, Inserm, Institut Pasteur de Lille, U1177–Drugs and Molecules for Living, Lille (France)
(c) Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm (Sweden)
(d) National Reference Center for Tuberculosis and Mycobacteria, Bacterial, Scientific Institute of Public Health (WIV-ISP), Brussels (Belgium)
(e) Tuberculosis Research Laboratory, Institut Pasteur Korea, (South Korea)
(f) Bioversys AG, Basel (Switzerland)
(g) Biozentrum, University of Basel, Basel (Switzerland)
(h) Laboratoire des Biopolymères et des Nanomatériaux Supramoléculaires, Université Libre de Bruxelles, Brussels (Belgium)
(i) VIB Center for Structural Biology, VIB, Brussels (Belgium)
(j) Structural Biology, Brussels, Vrije Universiteit Brussel (VUB), Brussels (Belgium),
(k) Swiss Tropical and Public Health Institute, Basel (Switzerland)
(l) University of Basel, Basel (Switzerland)
References
[1] C. Walsh, Antibiotics: Actions, Origins, Resistance (ASM Press, 2003)
[2] C. Reading and M. Cole, Antimicrob. Agents Chemother. 11, 852–857 (1977)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.