- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Dynamics and extreme conditions
- RXES investigation of d-d states in a self-aggregating supramolecular Pt-tpy complex
RXES investigation of d-d states in a self-aggregating supramolecular Pt-tpy complex
In the photophysics and photochemistry of transition metal complexes d–d (ligand field), transitions are of fundamental importance because they are often responsible for deactivating radiative (emissive) relaxation pathways to the ground state and for promoting ligand release photochemical reactions [1]. Despite their fundamental importance, it is difficult to characterise such states since they are spectroscopically silent to most conventional laboratory techniques. Nevertheless, it has been demonstrated in several cases that RXES (Resonant X-ray Emission Spectroscopy) can effectively provide specific information on d–d states for metal-containing systems. RXES is an element selective technique which probes both the unoccupied and occupied orbitals involved in electronic transitions at the metal centre by combining XANES and XES (Figure 43).
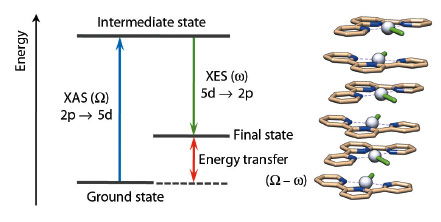 |
|
Fig. 43: Simplified energy diagram for RXES at the Pt L edge and example of stacked structure for [Pt(tpy)Cl]Cl. Colour code: Grey = Pt, light brown = C, blue = N, and green (stick) = Cl. |
At beamline ID26, we have extended the use of RXES to the study of d–d states in the self-aggregating square planar [Pt(tpy)Cl]Cl complex. In solution Pt-tpy (tpy = 2,2’:6’,2”-terpyridine) derivatives form oligomers of various sizes depending on their concentration (see for example Figure 43 where a model stacked structure is shown). The intermolecular Pt···Pt and π–π interactions that drive the formation of these supramolecular structures in polar solvents produce dramatic changes in their absorption and emission profile, as for example a luminescence shift to the near infrared. For these attractive optical features, Pt-tpy complexes have found application as sensors for biomolecules (e.g. glucose) and as probes for DNA G-quadruplex formation [2].
Collection of 2D RXES maps for [Pt(tpy)Cl]Cl at the Pt-L3 edge was achieved both on a solid sample (Figure 44) and on an aqueous solution. Aided by DFT calculations, simulation of the maps evidenced the nature of the orbitals involved in the d–d transitions. The main peak (EI = 11567.7 eV, ET = 5.1 eV) and the shoulder (ET = 6.7 eV) arise from Pt-based molecular orbitals with prevalent Pt 5d and Cl 3p character, while the tail (ca. ET = 7.5 eV) is attributed to transitions between molecular orbitals where the Pt 5d atomic orbitals are mostly mixed with N 2p and C 2p atomic orbitals of the tpy ligand.
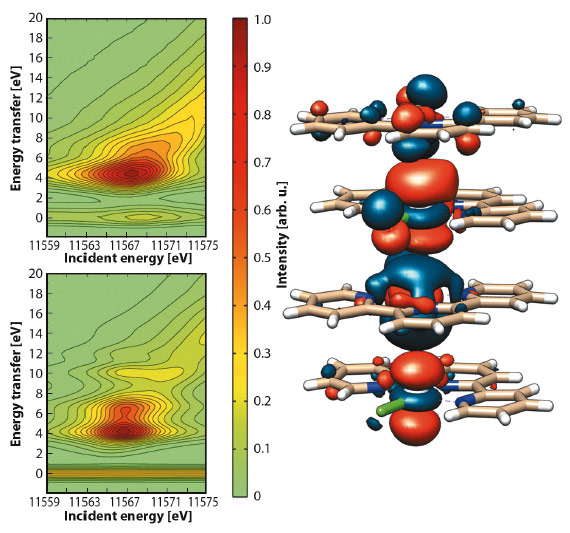 |
|
Fig. 44: Contour plots of experimental (top) and simulated (bottom) Pt-L3 (2p3/2) RXES planes for solid [Pt(tpy)Cl]Cl and selected antibonding molecular orbital for {[Pt(tpy)Cl]+}4. |
Notably, measurements performed on [Pt(tpy)Cl]Cl aqueous solutions of different concentrations (5–40 mM) retain these RXES features, but show a shift of the major peak to higher energy transfer values (up to 0.5 eV) as the sample concentration decreases. Such significant change in energy is associated with the concentration-dependent size decrease of the supramolecular structure of [Pt(tpy)Cl]Cl in diluted solutions. The filled d orbitals that extend perpendicular to the molecular plane (dxz, dyz and dz2) interact along the axial direction, splitting into bonding and antibonding combinations which lower the energy of d–d ligand field transitions. This is reflected in the shift of the RXES main band to higher energies at lower concentration.
Our findings help to rationalise the considerable changes in the optical properties of Pt-tpy derivatives with the substitution of a Cl ligand in [Pt(tpy)Cl]Cl and suggests that favouring the formation of supramolecular interactions in the plane of the ligands allows full control of the d–d states to be achieved by directly affecting the dx2–y2 orbital energy.
For the first time we demonstrated how this powerful technique can be used to characterise spectroscopically d-d states in high-order metal-based systems in solution. A complete understanding of the energy and orbital nature of these states is key to guide the design and synthesis of functional supramolecular architectures based on transition metal complexes.
Principal publication and authors
C. Garino (a), E. Gallo (a,b), N. Smolentsev (b,c), P. Glatzel (b), R. Gobetto (a), C. Lamberti (a), P.J. Sadler (d) and L. Salassa (d,e), Phys. Chem. Chem. Phys. 14, 15278-15281 (2012).
(a) Department of Chemistry and NIS Centre of Excellence, University of Turin (Italy)
(b) ESRF
(c) Research Center for Nanoscale Structure of Matter, Southern Federal University, Rostov-on-Don (Russia)
(d) Department of Chemistry, University of Warwick, Coventry (UK)
(e) CIC biomaGUNE, Donostia-San Sebastian (Spain)
References
[1] P.S. Wagenknecht and P.C. Ford, Coord. Chem. Rev. 255, 591-616 (2011).
[2] V.W.-W. Yam and K.M.-C. Wong, Chem. Commun. 47, 11579-11592 (2011).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.