- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2016
- Matter at extremes
- New type of charge-ordering transition in the novel iron oxide Fe4O5
New type of charge-ordering transition in the novel iron oxide Fe4O5
Synthesis of new classes of compounds can lead to discoveries of novel physical phenomena as well as innovative applications. Fe4O5 is the first member of a very recently discovered family of iron oxides that can be synthesised only under high-pressure conditions. A multi-technique study of Fe4O5 reveals that it undergoes a unique charge-ordering transition below 150 K that involves competing dimeric and trimeric ordering within the chains of Fe ions. This electronic transition drives an intricate incommensurate distortion in its crystal structure.
At ambient pressure, only three iron oxide polymophs were known, FeO, Fe3O4, and Fe2O3. Recently, several new iron oxide polymorphs with hitherto unknown stoichiometries have been discovered under high pressure conditions [1,2]. Among them, Fe4O5 looks particularly exciting as it can be readily quenched at ambient pressure [1]. We have performed a series of low-temperature studies on a recently-discovered high-pressure polymorph of iron oxide, Fe4O5 [1] and discovered a new type of charge-ordering transition involving the formation of competing dimeric and trimeric ordering within the chains of Fe ions, as revealed by X-ray diffraction studies at beamline BM01, SNBL. These findings were supported in neutron diffraction experiments and in measurements of magnetic and transport properties. To date, such exotic transitions have never been observed, and, hence, it brings new perspectives on charge-ordered states and transitions.
 |
|
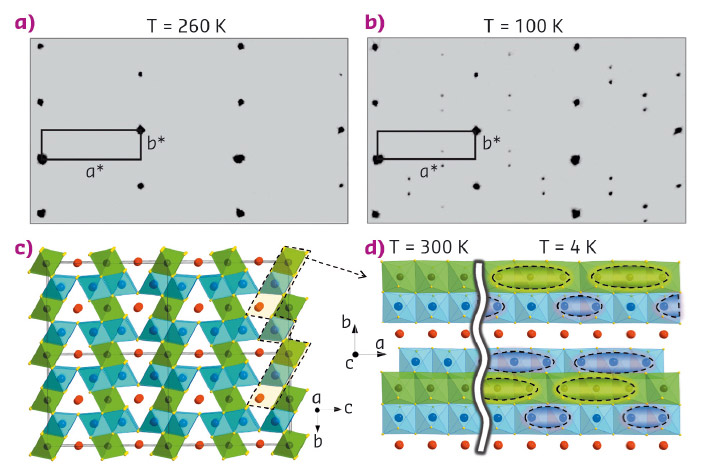
Fig. 129: (a, b) Examples of reciprocal lattices of X-ray diffraction intensities of Fe4O5 at 260 K and 100 K (c) Crystal structure projected down the a-axis at room temperature. d) Crystal structure of Fe4O5 projected along the c-axis at room and low temperatures. |
At ambient conditions, Fe4O5 crystallises in a CaFe3O5-type structure featuring linear chains of octahedrally-coordinated iron ions and linear chains of FeO6 trigonal prisms along the a-axis (Figure 129a,c). This compound contains equal amounts of Fe2+ and Fe3+ ions, and like another mixed-valent iron oxide, magnetite (Fe3O4), it is a good electrical conductor owing to a charge transfer between Fe2+ (charges) and Fe3+ (vacancies). Magnetite is known to undergo a charge-ordering phase transition below 125 K, which is accompanied by a jump in electrical resistivity [3]. This transition in magnetite had been discovered by Verwey in 1939 [3], but only recently by means of single-crystal X-ray diffraction, the charge-ordering pattern in its low-temperature phase has finally been uncovered to involve 'three-site-distortions', called 'trimerons' [4]. Analysis of the bond valence sums in the lattice of Fe4O5 indicated the mixed valence states of iron at the octahedrally-coordinated sites (Figure 129c), and, hence, one could expect that Fe4O5 also undergoes some charge ordering at relatively low temperatures.
 |
|
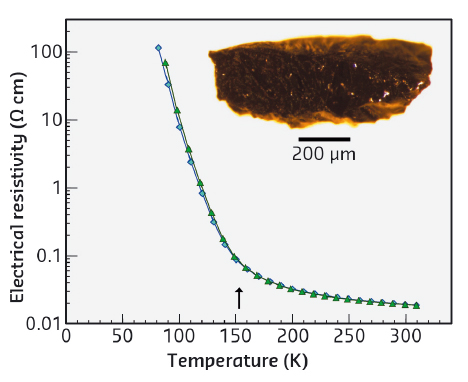
Fig. 130: Temperature dependencies of electrical resistivity of Fe4O5 in 0 and 12 T magnetic fields (blue and green curves, respectively). These curves exhibit a bend at 150 K (marked by the arrow), indicating a 'metal-insulator'-type transition. A photograph of the Fe4O5 sample is included. |
The high-quality single crystal X-ray diffraction patterns of Fe4O5 collected at beamline BM01 demonstrated the appearance of superlattice reflections upon cooling below 150 K (Figure 129a,b). Analysis of these patterns revealed that the low-temperature structure is incommensurately modulated and contains the Fe dimers and trimers within the chains of the octahedrally-coordinated iron (Figure 129d). A constant Fe-Fe distance of 2.861 Å (at 4 K) in the trigonal chains of ferrous iron enabled the dramatic shortening of some interoctahedra distances (marked by elongated ellipsoids in Figure 129d) to be highlighted. Each chain of the octahedral cations contains either dimers composed of Fe2+–Fe3+ pairs with one shared electron, or trimers composed of one Fe2+ and two Fe3+ ions, similar to the trimerons in Fe3O4 [4] (Figure 129d). This unusual charge-ordering transition in Fe4O5 is concurrent with a significant increase in electrical resistivity (Figure 130), and therefore, it may be classified as a "metal-insulator"-type transition. The magnetic susceptibility measurements and neutron diffraction establish the formation of a collinear antiferromagnetic order above room temperature and a spin canting at 85 K that gives rise to spontaneous magnetisation.
Principal publication and authors
Charge ordering transition in iron oxide Fe4O5 involving competing dimer and trimer formation, S.V. Ovsyannikov (a), M. Bykov (a,b), E. Bykova (a,b), D.P. Kozlenko (c), A.A. Tsirlin (d,e), A.E. Karkin (f), V.V. Shchennikov (f,g), S.E. Kichanov (c), H. Gou (a,h), A.M. Abakumov (i,j), R. Egoavil (i), J. Verbeeck (i), C. McCammon (a), V. Dyadkin (k), D. Chernyshov (k), S. van Smaalen (b) and L.S. Dubrovinsky (a), Nature Chemistry 8, 501-508 (2016); doi: 10.1038/nchem.2478.
(a) Bayerisches Geoinstitut, Universität Bayreuth (Germany)
(b) Laboratory of Crystallography, Universität Bayreuth (Germany)
(c) Frank Laboratory of Neutron Physics, JINR, Dubna (Russia)
(d) National Institute of Chemical Physics and Biophysics, Tallinn (Estonia)
(e) Institute of Physics, University of Augsburg (Germany)
(f) Institute of Metal Physics, Russian Academy of Sciences, Yekaterinburg (Russia)
(g) Institute for Solid State Chemistry, Russian Academy of Sciences, Yekaterinburg (Russia)
(h) Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao (China)
(i) Electron Microscopy for Materials Research (EMAT), University of Antwerp (Belgium)
(j) Department of Chemistry, Moscow State University (Russia)
(k) Swiss–Norwegian Beamlines at the ESRF, Grenoble (France)
References
[1] B. Lavina, et al. Proc Nat. Acad. Sci. US 108, 17281 (2011).
[2] E. Bykova, et al., Nature Commun. 7, 10661 (2016).
[3] E.J.W. Verwey, Nature 144, 327 (1939).
[4] M.S. Senn et al., Nature 481, 173 (2012).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.