- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2011
- Electronic structure and magnetism
- The elusive structure and symmetry of the proton conducting oxide Y:BaZrO3
The elusive structure and symmetry of the proton conducting oxide Y:BaZrO3
Proton-conducting oxides are the most studied electroceramics for hydrogen technology such as fuel cells, membranes for hydrogen purification and hydrogen sensors [1]. Their advantage over zirconia electrolytes for fuel cells relies on the lower operating temperature range (500-700°C). Among proton-conducting perovskite oxides, yttrium-doped barium zirconate BaZr1-xYxO3-y (BZY) has been proposed as the best candidate for technological applications, combining both high proton mobility and chemical stability.
In the last decade, many crystal structures have been proposed for BZY compounds using X-ray or neutron diffraction: cubic, tetragonal, orthorhombic, or an intrinsically two-phase regime with different cation compositions. Moreover, the yttrium dopant atoms are a potential source of local distortions, or even of preferential attraction for protonic defects: in this respect, the local structure approach has been overlooked in the past, and only recently applied to proton-conducting perovskites [2].
To unravel the finer structural details of BZY, we used X-ray diffraction, exploiting the exceptional resolution of beamline ID31 to probe long-range symmetry, and X-ray absorption spectroscopy at beamline BM29, to explore the local structure around zirconium and yttrium: these were complemented with Raman spectroscopy to probe the local symmetry.
We have determined that BZY forms a single crystalline phase for Y3+ content up to 15%. If probed with XRD, undoped BaZrO3 shows a perfectly cubic perovskite structure, but the high oxygen vibration amplitudes and the Raman peaks suggest that incoherently mutual tilting of ZrO6 units appears throughout the lattice (not affecting the overall cubic symmetry). This dynamical distortion of the anion sublattice is an unexpected and interesting feature, possibly present also in other cubic perovskites.
 |
|
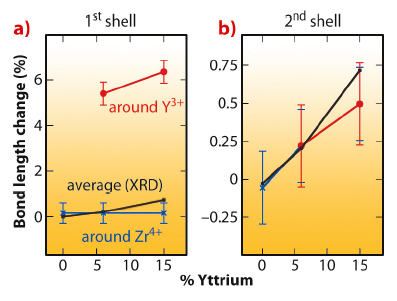
Fig. 93: Interatomic distances of first and second shell around different sites as a function of Y3+ dopant content, represented as bond length change with respect to undoped BaZrO3. a) First shell distances Y-O (red), Zr-O (blue) and average M-O (black), as derived by both EXAFS and XRD analysis. b) Y-Ba (red), Zr-Ba (blue) and average M-Ba (black). |
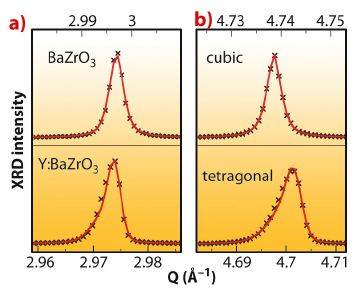
When Y3+ dopants are inserted in the perovskite lattice they act as rigid inclusions, maintaining their larger ionic radius with respect to the host Zr4+ cations. However, the cation and anion sublattices respond very differently to provide sufficient space for the YO6 octahedra: the O2- shells are much expanded around the dopant, while the Y-Ba and Zr-Ba bond lengths are equal (see Figure 93), like Y-Zr and Zr-Zr. This peculiar configuration is attained though a local tilting of the YO6 units with respect to the rest of the lattice, resulting in local Y-O-Zr configurations with angles of about 160°. The tilting of the YO6 octahedra causes a reduction of symmetry throughout the structure, with an extremely slight tetragonal distortion (< 0.1%) appearing, as proved by the splitting of many XRD peaks (see Figure 94).
 |
|
Fig. 94: XRD data (black crosses) and Rietveld refinement (red line) of undoped BaZrO3 and BZY with 15% Y3+. a) The (310) reflection of the cubic cell splits into (113) and (331) reflections in the case of a tetragonal cell. b) The (200) reflection of the cubic cell splits into (002) and (220) reflections in the case of a tetragonal cell. |
These structural distortions around Y3+ are most likely the reason of its limited solubility in the BaZrO3 lattice. However, they are not large enough to create two inequivalent oxygen sites, and therefore not reducing the possible hopping pathways for protons.
Principal publication and authors
F. Giannici (a), M. Shirpour (b), A. Longo (c), A. Martorana (a), R. Merkle (b) and J. Maier (b), Chem. Mater. 23, 2994-3002 (2011).
(a) Università di Palermo, Palermo (Italy)
(b) Max-Planck-Institut für Festkörperforschung, Stuttgart (Germany)
(c) Istituto per lo Studio dei Materiali Nanostrutturati, CNR, Palermo (Italy)
References
[1] K.-D. Kreuer, Annu. Rev. Mater. Res. 33, 333-359 (2003).
[2] F. Giannici, A. Longo, A. Balerna, K.-D. Kreuer and A. Martorana, Chem. Mater. 19, 5714-5720 (2007).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.