- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2009
- Structural biology
- Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2
Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2
The ribosome – the cellular machinery responsible for protein synthesis – reads the codons (triplets of bases coding for amino acid residues) of messenger RNAs to assemble polypeptide chains. A particular triplet, the stop codon, signals to the ribosome the end of the coding region and triggers translation termination. A mutagenic event (e.g. a genetic mutation) can randomly introduce an abnormal stop codon (nonsense mutation) into the coding region of a messenger RNA. This would cause premature translation termination and thus the production of a truncated protein. Such aberrant proteins often display severe toxic effects on cells and are responsible for several diseases like thalassemia and cystic fibrosis.
Eukaryotic cells have evolved a defense mechanism against such mutations called nonsense-mediated mRNA decay (NMD) [1]. A fundamental role in NMD is carried out by the exon junction complex (EJC), a multi-protein complex deposited by the splicing machinery onto the mRNA just upstream from each exon-exon junction. The ribosome stalled at a premature termination codon recruits other factors – notably the NMD protein UPF1 – and forms the so-called SURF complex (named after the component proteins SMG1, UPF1 and eRF1-3). Unless the premature termination codon is found in the last exon, an EJC complex is found downstream of the SURF complex. The NMD factor UPF2, associated with the EJC through its interaction with UPF3, recognises and binds UPF1 on the SURF complex thus bridging the two macromolecular complexes. This key recognition triggers a series of downstream events that finally lead to the degradation of the mRNA, protecting the cell from the production of a potentially dangerous truncated protein.
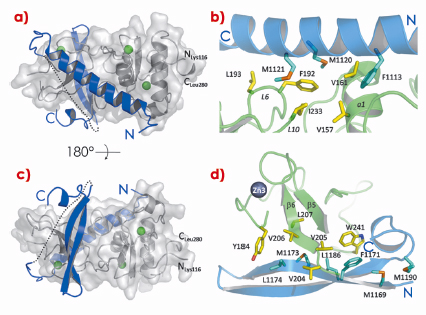 |
|
Fig. 119: a), b) UPF2 (blue) is represented as ribbons and UPF1 (grey) as ribbons and a transparent surface. UPF1 zinc atoms are shown in green. The UPF2 missing linker is represented as a dotted line. c) The principal residues of the UPF2 N-terminal helix (cyan) and the UPF1 CH domain (yellow) that form the hydrophobic interface between the two molecules are represented as sticks. d) The main interacting residues of UPF2 C-terminal b-hairpin (cyan) and UPF1 CH domain (yellow) are represented as sticks. |
Here we report structural studies on the unusual interaction between the C-terminal region of UPF2 and UPF1. UPF1 is an RNA helicase with an additional N-terminal Cys/His-rich domain (CH domain). The crystal structure of the UPF1-UPF2 complex (obtained at beamline ID14-4) shows that UPF2 adopts an unusual bipartite mode of interaction with UPF1, binding via separated alpha-helical and beta-hairpin elements connected by a long linker on opposite sides of the CH-domain of UPF1 (Figure 119). Complementary NMR and ITC measurements show that the interacting region of UPF2 is intrinsically disordered in the unbound state and folds upon binding to UPF1, with the beta-hairpin element having the highest affinity. An additional crystal structure (obtained at beamline ID29) shows that UPF1 CH domain is docked onto the helicase in a fixed configuration even in the absence of UPF2. Small-angle X-ray scattering (SAXS) measurements performed on the BioSAXS beamline ID14-3 confirmed that the relative orientation of the two domains is maintained in solution independently of bound UPF2 (Figure 120).
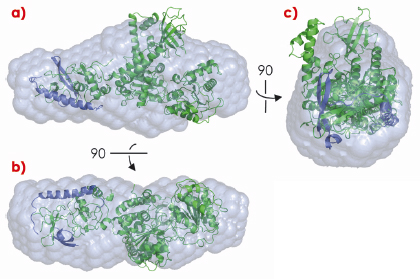 |
|
Fig. 120: a) Side, b) bottom and c) front views of the crystal structure of UPF1 in complex with the C-terminal domain of UPF2, superposed on the corresponding ab initio model calculated from the SAXS scattering curve. |
In summary, we show that the UPF1-binding region of UPF2 shares the typical features of intrinsically disordered regions, which are known to mediate very specific and often transient interactions. We propose that UPF2 uses its disordered C-terminal region as a long, flexible “fishing line” to allow rapid recognition and binding of UPF1, the co-folding upon binding ensuring high specificity. This mechanism is appropriate as, by virtue of its flexibility, the UPF2 fishing line can explore large volumes and overcome topological constraints to ensure that the key connection is made between the EJC and UPF1 on the stalled ribosome, thus leading to efficient degradation of premature termination codon-containing aberrant mRNAs.
Reference
[1] G. Neu-Yilik and A.E. Kulozik, Adv Genet 62, 185-243 (2008).
Principal publication and authors
M. Clerici (a,b), A. Mourão (c,d,e), I. Gutsche (b), N.H. Gehring (f,g), M.W. Hentze (d,f), A.E. Kulozik (f,g), J. Kadlec (a,b), M. Sattler (c,e) and S. Cusack (a,b), Embo J 28, 2293 – 2306 (2008).
(a) European Molecular Biology Laboratory, Grenoble Outstation (France)
(b) Unit of Virus Host-Cell Interactions, UJF-EMBL-CNRS (France)
(c) Munich Center for Integrated Protein Science, Technische Universität (Germany)
(d) European Molecular Biology Laboratory, Heidelberg (Germany)
(e) Institute of Structural Biology, Helmholtz Zentrum München (Germany)
(f) Molecular Medicine Partnership Unit, EMBL and University of Heidelberg (Germany)
(g) Department of Pediatric Oncology, Children’s Hospital, University of Heidelberg (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.