- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2010
- Structure of materials
- Structure and energetics of azobenzene on Ag(111): Benchmarking semiempirical dispersion correction approaches
Structure and energetics of azobenzene on Ag(111): Benchmarking semiempirical dispersion correction approaches
Functional organic molecules at metal surfaces are studied because of their potential as a future molecular nanotechnology. A simulation platform is required that would allow the precise prediction of all relevant modes of the molecule-substrate interactions (chemical bonds of specific groups, Pauli repulsion, steric hindrance, and dispersion interaction). For this, the adsorption of organic molecules with highly polarisable conjugated ring systems on metals poses particular difficulties because the prevalence of dispersive van der Waals (vdW) interactions restricts the applicability of semilocal exchange and correlation (xc) functionals within density-functional theory (DFT). Since high-level theories that include nonlocal vdW interactions by construction are still barely tractable for large surface-adsorbed molecules, computationally inexpensive semi-empirical dispersion correction schemes to semi-local DFT (DFT-D) represent an appealing alternative. In these approaches, vdW interactions are considered approximately by adding a pairwise interatomic C6R-6 term to the DFT energy. At distances below a cutoff, specified via the vdW radii of the atom pair, this long-range dispersion contribution is heuristically reduced to zero by multiplication with a short-range damping function.
The applicability of this DFT-D approach to organic molecules adsorbed at metal surfaces is uncertain. It is not clear how the results are affected by neglecting the screening of dispersive interaction between the adsorbate and more distant substrate atoms by the intervening metal layers. Moreover, adsorbate molecules, which also interact covalently with the substrate, exhibit bond distances that are so short that the uncertainties in the heuristic damping function of the dispersion term might mingle in an uncontrolled way with deficiencies of the employed semilocal DFT functional. Therefore, accurate experimental reference data are required to determine whether the improvements brought by including vdW interactions semi-empirically outweigh the aforementioned shortcomings.
 |
|
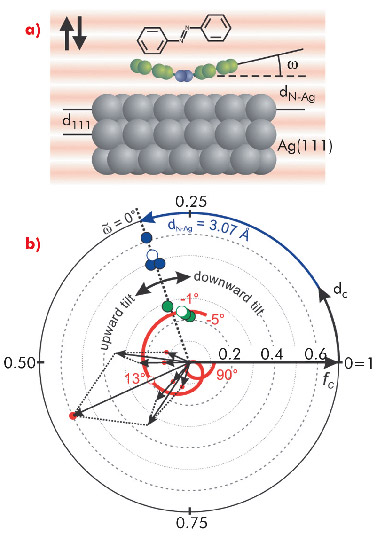
Fig. 34: a) Schematic adsorption model of azobenzene on Ag(111) in the X-ray standing wave field; b) Argand diagram of experimental coherent position dc and fraction fc for N1s (blue circles) and C1s (green circles). The red spiral represents the calculated dc and fc for C1s as w sweeps from -5° to 90°; its intersection with experimental C1s data points defines the tilt angle of the phenyl rings. |
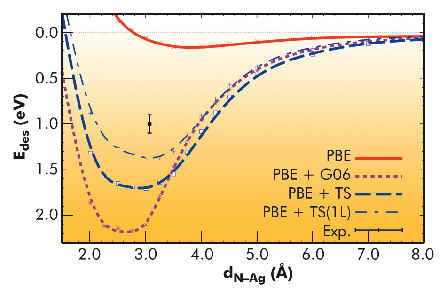
We respond to this need by providing reference data for the molecular switch azobenzene on Ag(111) (Figure 34). The normal-incidence X-ray standing wave technique (NIXSW) at beamline ID32 was used to determine key structural parameters of adsorbed azobenzene. Additionally, the desorption energy was measured by temperature programmed desorption (TPD). Employing a novel analysis scheme for NIXSW (Figure 34b), we extracted not only the height of azobenzene diazo-bridge dN-Ag that is straightforwardly accessible in NIXSW, but also the tilt of the phenyl rings ω. The derived structural data (dN-Ag = 3.07±0.02 Å and ω = -1±1.5°) demonstrate an essentially undistorted planar molecular geometry and agree well with the results obtained with DFT-D scheme due to Tkachenko and Scheffler (TS) [1]. Indeed, while the DFT-PBE strongly overestimates the diazo-bridge adsorption distance (3.64 Å) demonstrating the well-known incapability of semilocal xc functionals to account for long-range dispersive interactions, the tested DFT-D schemes by Grimme (G06) [2] and TS reveal a sizable reduction of dN-Ag to 2.75 and 2.98 Å (Figure 35). All three schemes predict almost flat molecular geometry in similar agreement with experiment.
Unfortunately, the improved structure in DFT-D goes together with a notable overbinding. Most probably the reason is the screening of dispersive attractions between the adsorbate atoms and more distant substrate atoms that is neglected in the strictly pairwise evaluation of the dispersion interaction as inspired by Hamaker theory. Mimicking this screening by reducing the number of substrate layers considered in the pairwise interaction C6R-6 term to one and by even diminishing the C6 coefficients of the Ag atoms in the topmost surface layer indeed shifts the computed binding energy curves closer to experimental values, but essentially without affecting the dN-Ag (Figure 35).
 |
|
Fig. 35: Computed desorption energy curves and experimental point. |
In summary, we demonstrate that the existing DFT-D schemes are not suitable to describe comprehensively the role of vdW interactions for adsorption at metal surfaces because screening effects are not (yet) considered. However, the insight that the adsorption geometries are less sensitive to the neglect of screening is intriguing. It suggests that these schemes may provide significantly improved structural data at zero additional computational cost. Such structures are then a useful starting point for higher-level theory, aimed at refined binding energies or a more comprehensive understanding of the adsorption system.
Principal publication and authors
G. Mercurio (a), E.R. McNellis (b), I. Martin (c), S. Hagen (b,c), F. Leyssner (b), S. Soubatch (a), J. Meyer (b), M. Wolf (b,c), P. Tegeder (c), F.S. Tautz (a) and K. Reuter (b,d), Phys. Rev. Lett. 104, 036102 (2010).
(a) Forschungszentrum Jülich (Germany)
(b) Fritz-Haber-Institut, Berlin (Germany)
(c) Freie Universität Berlin (Germany)
(d) Technische Universität München (Germany)
References
[1] A. Tkatchenko and M. Scheffler, PRL 102, 073005 (2009).
[2] S. Grimme, J. Comput. Chem. 27, 1787 (2006).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.