- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2000
- Chemistry
- Picosecond Photochemistry in Solution
Picosecond Photochemistry in Solution
Femtosecond spectroscopy has now reached a high level of maturity. However, one intrinsic limitation of the technique is that when interpreting spectroscopic results the energy surfaces of the ground state and all excited species must be known in detail. For this reason, as well as the influence of thermal effects and impurities, the interpretation of a spectroscopic signal rapidly becomes challenging as the system of study increases in complexity. Since X-rays have a wavelength of the order of atomic spacings, a diffuse X-ray scattering pump-probe approach offers the promise of visualising, in real time, rapid structural changes in photochemical systems [1]. This potential was recognised by Professor Ahmed Zewail (1999, Nobel Prize in Chemistry), who has developed a picosecond pump-probe methodology using electron diffraction from a molecular beam in a vacuum environment [2]. A number of technical challenges are posed by picosecond diffuse X-ray scattering. There is no crystal lattice with which to amplify the signal above the background and, at typical concentrations, tens of millimolar, only one molecule in a thousand is the photochemical species of interest. Hence the experimental strategy must contend with challenging signal-to-noise considerations, making the peak X-ray brilliance available at the ESRF a prerequisite.
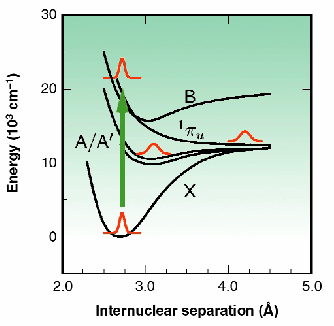 |
Fig. 12: Schematic illustration of the energy surfaces of molecular iodine.
|
Proof-of-principle experiments were performed at ID9 using iodine in dichloromethane as a test system. The energy-level surfaces of this system are illustrated in Figure 12. Following photo-excitation to a repulsive energy surface, the two iodine atoms move apart. Within a few picoseconds their kinetic energy becomes absorbed by the surrounding solvent molecules. As such, the iodine atoms may recombine directly onto the X surface, or recombine first onto the A/A' surface, or may escape the solvent cage altogether. In dichloromethane, the A/A' state has a lifetime of 500 ps, suitable for study using the 80 ps synchrotron X-ray pulse duration. Figure 13a shows the experimentally measured changes in the diffuse X-ray scattering intensities following sample photo-excitation by a fs light pulse. An oscillatory behaviour is observed, illustrating a decreased scattering intensity at low resolution, and a corresponding increase at medium deflection angle. Overlaid on this data is the theoretically expected curve when the photolysed sample has a population of 25% A/A' and 5% escapes the solvent cage. An oscillation at higher deflection angle was not resolved, possibly because of thermal effects perturbing the solvent atoms in a random way and washing out this feature. Figure 13b shows the variations in the signal with time measured using a gas-filled detector coupled to a lock-in amplifier. The expected temporal response of the system is shown as a solid line. Because of the small amplitude of the desired signal, improvements in the experimental approach seek to maximise the X-ray flux through the sample. As such, a move to a narrow "pink- Laue" undulator line, or multi-layer wide-bandwidth X-ray optics, point the way towards the study of increasingly complex systems.
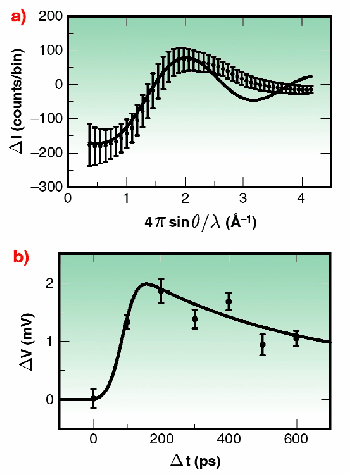 |
Fig. 13: a) Experimentally measured changes (photon counts per 100 pixel bin) resulting from the photolysis of 40 mMol I 2 in CH 2Cl 2. The solid line shows the predicted change. b) Temporal dependence of the difference signal due to the photolysis of I 2 in CH 2Cl 2. Each data point records the relative amplitude of the negative peak of Figure 13a sampled from Q = 0.4 Å -1 to 1.5 Å -1. The response of the system was predicted (solid line) from the 500 ps lifetime of the A/A' state and a constant off-set due to cage break-out.
|
References
[1] J.P. Bergsma, M.H. Coladonato, P.M. Edelsten, J.D. Kahn, K.R. Wilson and D.R. Fredkin, J. Chem. Phys., 84, 6151-6160 (1986).
[2] J.C. Williamson, J.Cao, H. Ihee, H. Frey and A.H. Zewail, Nature, 386, 159-162 (1997).
Authors
R. Neutze (a), R. Wouts (a), S. Techert (b), J. Davidsson (a), M. Kocsis (b), A. Kirrander (a), F. Schotte (b) and M. Wulff (b).
(a) Uppsala University (Sweden)
(b) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.