- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Matter at extremes
- Redox processes in Fe catalysts resolved by XAS and UV/Vis spectroscopy
Redox processes in Fe catalysts resolved by XAS and UV/Vis spectroscopy
The iron oxidation state and local structure in a bioinspired non-heme-based catalyst used in C-H and C=C oxidation reactions was studied using energy dispersive XAS and UV/Vis spectroscopy to follow the reactions on a millisecond timescale and identify the intermediate species.
Aerobic life has evolved to depend on the oxidising power of O2 in various metabolic pathways. Enzymes, such as non-heme iron-dependent oxygenases, catalyse the incorporation of O2 into a wide range of biological molecules and use diverse strategies to activate their substrates. One way for C-H activation to occur is by the hydroxylation of the C-H bond. Bioinspired non-heme aminopyridine iron complexes/H2O2 catalytic systems replicate the oxidation mechanism of the elaborate non-heme iron oxygenases with simple iron coordination complexes. The key feature of these mechanisms is a controlled O-O bond activation that does not generate free-diffusing radical intermediates (which undermine the oxidation selectivity). This pathway is facilitated by the different oxidation states taken by iron during the catalytic cycle (from FeII up to FeIV or FeV). However, reaction conditions have great influence on the mechanistic pathway and the key intermediates are elusive to conventional spectroscopies.
 |
|
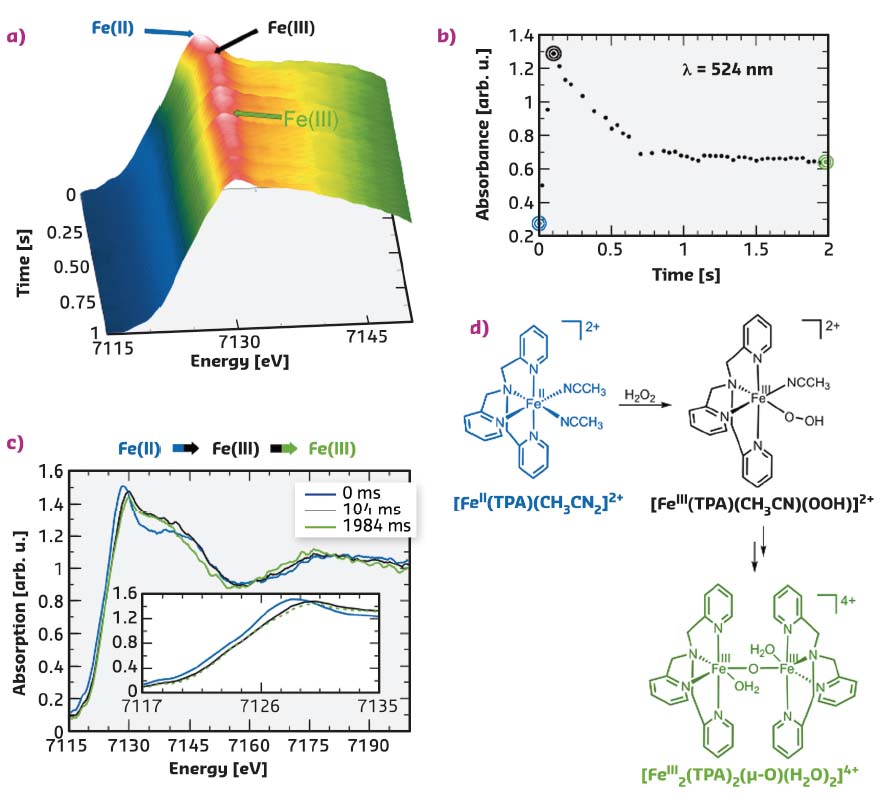
Fig. 37: a) Time evolution of the Fe K-edge EDXAS spectra of [FeII(TPA)(CH3CN)2]2+ (35 mM) after addition of H2O2 (70 mM) in CH3CN/H2O 99.8:0.2 (v/v) at 25°C. b) Time evolution of the absorbance at λ = 524 nm of the same reaction mixture. c) Selected EDXAS spectra including an enlargement of the Fe K-edge region. d) Sequence of iron species compatible with the EDXAS-UV/Vis spectral evolution. |
FeII(tris(2-pyridylmethyl)amine), [FeII(TPA)(CH3CN)2]2+ is a paradigmatic example of such non-heme iron complexes, and here its reaction with different oxidants has been studied with a simultaneous EDXAS-UV/Vis approach at beamline ID24. Figure 37a shows the evolution of the EDXAS spectra during the course of the reaction of [FeII(TPA)(CH3CN)2]2+ (35 mM) with H2O2 (70 mM) at 25°C. The XANES spectra show both a rapid shift in the Fe K-edge position within the first 104 ms and a modification of the structural oscillations, in accordance with an initial oxidation of FeII to FeIII followed by a clear rearrangement of the local structure around the metal centre, without further changes in the iron oxidation state. Figure 37b shows the absorbance evolution at λ = 524 nm recorded simultaneously. The increase in absorbance at this wavelength is consistent with the formation of the [FeIII(TPA (CH3CN)(OOH)]2+ species (λmax = 538 nm). The subsequent absorbance decrease occurring between 104 and 704 ms illustrates the evolution of this intermediate to the μ-oxo dimer [FeIII2(TPA)2(µ-O (H2O)2]4+ as depicted in Figure 37. The XANES spectra collected at t = 0, 104, and 1984 ms are shown in Figure 37c. The EDXAS analysis confirms the observations made with UV/Vis and the experimental body of evidence indicates the reaction mechanism depicted in Figure 37d.
 |
|
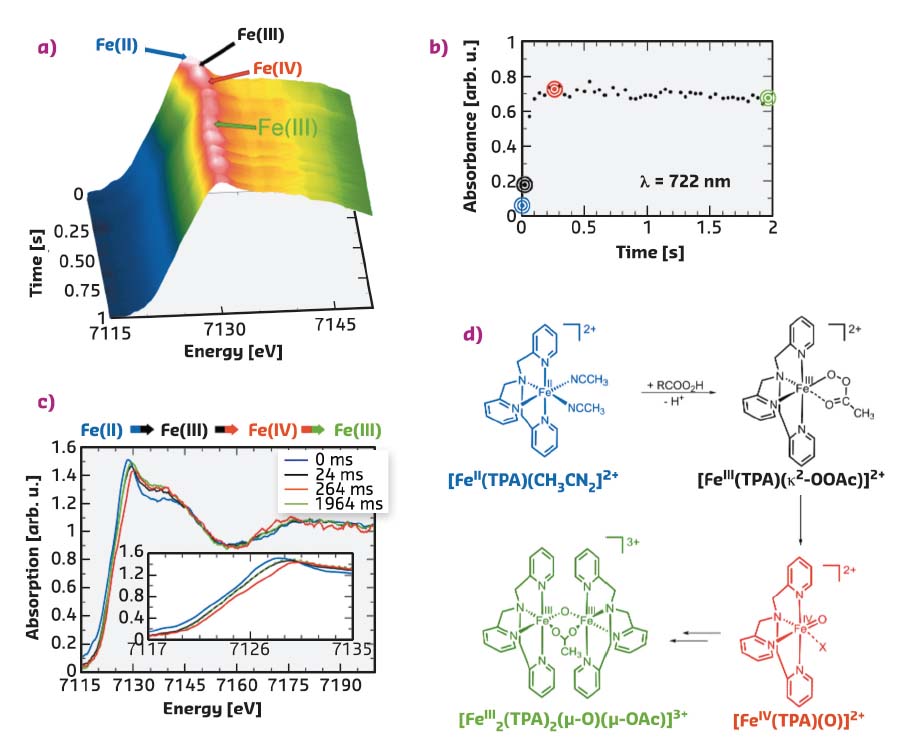
Fig. 38: a) Time evolution of the Fe K-edge EDXAS spectra of [FeII(TPA)(CH3CN)2]2+ (35 mM) after addition of CH3COO2H (35 mM) in CH3CN/CH3COOH 99.6:0.4 (v/v) at 25°C. b) Time evolution of the absorbance at λ = 722 nm. c) Selected EDXAS including an enlargement of the Fe K-edge region. d) Sequence of iron species compatible with the EDXAS-UV/Vis spectral evolution (X = OAc, CH3CN). |
A second reaction of [FeII(TPA)(CH3CN)2]2+ (35 mM) and CH3COO2H (35 mM) as the oxidant was carried out in CH3CN/CH3COOH 99.6:0.4 (v/v) at 25°C. Figure 38a shows the XANES spectra collected every 40 ms. The Fe K-edge position of the spectrum recorded after 24 ms is found to be shifted towards higher energy with respect to the initial [FeII(TPA)(CH3CN)2]2+ spectrum as evident from the selected XANES spectra reported in Figure 38c (compare the black and blue traces in the inset). Within the following 264 ms, the edge position is further shifted to higher energy, and then it moves back to lower energy during the subsequent 1700 ms. The time evolution of the Fe K-edge position is clearly visible in Figure 38c, where the XANES spectra collected at t = 0, 24, 264 and 1964 ms are reported. There is an edge shift of 0.7 eV in going from the [FeII(TPA)(CH3CN)2]2+ to [FeIII(TPA (κ2-OOAc)]2+ species and a further shift of 0.7 eV when the [FeIV(TPA)(O)]2+ species is formed. The absorbance at 722 nm has been monitored simultaneously and its time evolution is shown in Figure 38b. This wavelength was chosen since the [FeIV(TPA)(O)]2+ complex has a characteristic pale green chromophore at λmax = 720 nm, while none of its monomeric precursors show absorption in this region. A significant increase in the absorbance is evident in the first phase of the reaction (0-264 ms), while a slight decrease is observed starting from 264 ms. The observed experimental evidence strictly reproduces the mechanistic sequence of events depicted in Figure 38d.
The combination of energy-dispersive XAS and UV/Vis spectroscopy allowed a direct analysis of the evolution of both the oxidation states and the local structures of the Fe complexes studied here. This combined approach is a powerful tool for the characterisation of chemical reaction intermediates and will provide unique insights into reaction mechanisms involving transition metals.
Principal publication and authors
Following a chemical reaction on the millisecond time scale by simultaneous X-ray and UV/Vis spectroscopy, G. Olivo (a), A. Barbieri (a), V. Dantignana (a), F. Sessa (a), V. Migliorati (a), M. Monte (b), S. Pascarelli (b), T. Narayanan (b), O. Lanzalunga (a), S. Di Stefano (a) and P. D’Angelo (a), J. Phys. Chem. Lett. 8, 2958-2963 (2017); doi: 10.1021/acs.jpclett.7b01133.
(a) Department of Chemistry, University “La Sapienza”, Rome (Italy)
(b) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.