- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Electronic structure, magnetism and dynamics
- Niche chemical bonding model for actinides could be a general rule
Niche chemical bonding model for actinides could be a general rule
Scientists at Manchester, Lancaster, Nottingham, Dresden and Grenoble have published research reporting a surprise finding about the nature of lanthanide and actinide chemical bonding which suggests that an important – but previously considered niche – bonding model that controls chemical structure and reactivity may be a more general rule.
The arrangement of ligand groups at metal centres in metal complexes is crucial to determining their reactivity. For example, cis-platin is very effective at treating cancerous tumours whereas trans-platin is ineffective; both compounds have the same formula with a central platinum ion bonded to two chloride and two ammonia ligands all in the same plane in the shape of a cross, but in the former the chlorides are next to each other and in the latter they are opposite each other. The phenomenon that electronically controls to a large extent the arrangement of ligands at metal centres is known as the trans-influence (TI), which originates from the fact that some ligands bind to metals more strongly than others so ligands will avoid being opposite stronger donor ligands. This effect is observed right across the periodic table.
In contrast, at the bottom of the periodic table, things can be quite different, and for actinides a phenomenon known as the inverse-trans-influence (ITI) operates. Here, ligands that would normally do everything they could to avoid being opposite each other at a metal actually do everything they can to be oppositely disposed, and rather than this destabilising the ligands they actually mutually reinforce each other. However, the ITI was previously limited only to early actinides in their maximum or close-to-maximum oxidation states. A famous example of this is uranyl, UO22+, which is naturally prevalent in the environment and nuclear waste, and where the two oxygen atoms reside opposite each other and are bonded very strongly to uranium. This makes the uranyl moiety very important, but also the universal view that the ITI was a niche concept of limited applicability dominated because it was restricted to high oxidation state actinides like uranium.
So whether this phenomenon was a niche rule or hinted at a broader underpinning role, in an area that famously has few rules, had been a question that endured for decades.
One reason this question had been difficult to tackle was the lack of structurally comparable families of molecules from which to conduct comparative studies where lower oxidation states of metal ions could be investigated. To address this problem, the research team prepared a new family of molecules with C=M=C cores where the metal was cerium (a lanthanide), or thorium or uranium (actinides), where two carbon atoms that are known to be strong donors were forced to be opposite to one another either side of the metal (Figure 102).
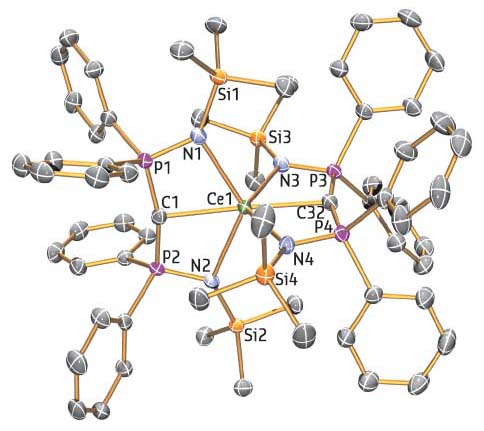 |
|
Fig. 102: Molecular structure of one of the key molecules in the study showing the linear C=M=C (M = cerium) core. |
Characterisation of this family of molecules required a multi-disciplinary effort, part of which was carried out at beamline BM20 (ROBL), combining structural, spectroscopic, and magnetic analyses to confirm the formulations of these molecules and computational chemistry to unravel their electronic structures. Surprisingly, this established that the ITI operates in these molecules that contain +4 oxidation state metal ions. The most telling observation is the fact that the two carbon atoms are bonded very closely to the metals, particularly for cerium, even though as strong donors they should be found at longer distances if the TI were operating, and this is a tell-tale sign of the presence of the ITI.
The combination of finding the ITI in +4 metal ion compounds for lanthanide as well as actinide complexes leads to the inescapable conclusion that this phenomenon is most likely of far broader reach than previously thought; if confirmed it would add a welcome rule to an area that famously lacks rules.
Principal publication and authors
The inverse-trans-influence in tetravalent lanthanide and actinide bis(carbene) complexes, M. Gregson (a), E. Lu (a), D.P. Mills (a), F. Tuna (b), E.J.L. McInnes (b), C. Hennig (c,d), A.C. Scheinost (c,d), J. McMaster (e), W. Lewis (e), A.J. Blake (e), A. Kerridge (f) and S.T. Liddle (a), Nature Communications 8, 14137 (2017); doi: 10.1038/ncomms14137.
(a) School of Chemistry, University of Manchester (UK)
(b) EPSRC National UK EPR Facility, School of Chemistry and Photon Science Institute, University of Manchester (UK)
(c) Helmholtz-Zentrum Dresden-Rossendorf, Institute of Resource Ecology, Dresden (Germany)
(d) ESRF
(e) School of Chemistry, University of Nottingham (UK)
(f) Department of Chemistry, Lancaster University (UK)
Acknowledgements
The project was also supported by the Royal Society, the Engineering and Physical Sciences Research Council, the European Research Council, The Universities of Manchester and Nottingham, the EPSRC UK National EPR Facility, the UK National Nuclear Laboratory, and the ESRF at Grenoble.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.