Data analysis
Data conversion and analysis
Data extraction
Further raw data treatment and summing of spectra
On-line fitting with minuit
Data conversion and analysis
On the data treatment/analysis computer brahe, the data directory of galilee is mounted directly (read only) with all your data in real time. In this way you can analyse the data on-line, without interfering with the data acquisition. Your local contact will create a directory where all the treated data as well as the results of the fits will be stored.
brahe:~% cd experiments
brahe:~/experiments% mkdir hsnnn
brahe:~/experiments% cd hsnnn
Data extraction
brahe:~/experiments/hsnnn% newplot
1. Using the file menu in newplot, go into the corresponding data directory of your experiment.
| or | data/id28/inhouse/galilee/runN_yy/hsnnn/expNN |
| /mntdirect/_data_id28/inhouse/galilee/runN_yy/hsnnn/expNN. |
2. Open the file which contains all the spectra.
You have a list of all your scans, you can visualise them, and make some basic analysis.
3. Set extraction script
Depending on the type of scan (energy scan, hkl-scan, or Q-scan) you have to choose the corresponding extraction script which extracts the relevant columns out of the data file, and performs the conversion of resistance (of the temperature sensors) into temperature.
-
Click on the button “Standard” on the bottom right part of the left panel and choose “Set Script”.
-
Go to the /users/opid28/usermacros/scriptextract and choose the appropriate script:
< tr> < tr> < tr>normextract : for standard IXS scans. hklextract: for Q-scans performed in hkl-mode (in the reciprocal lattice). qextract: for S(Q) scans
4. Selection of the scans to be extracted
When you have chosen which scans you are interested in, select them by clicking (left button) on the little arrow on the left of the scan name (or select them and click on the select button). Then push the extract button on the bottom. newplot then extracts the chosen scans into separate files expN.nscan (nscan= number of the scan), with the following columns:
If you use normextract:
I0, I1 , Tmono, 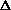 T1(ana1-mono), T2(ana2-mono), ..., 5, deta1, deta2, deat3, deat4, deta5
T1(ana1-mono), T2(ana2-mono), ..., 5, deta1, deta2, deat3, deat4, deta5
If you use hklextract:
H, K, L, 2![]() , I0, I1 , Tmono, T1(ana1-mono), T2(ana2-mono), ..., T5, deta1, deta2, deat3, deat4, deta5, detsq
, I0, I1 , Tmono, T1(ana1-mono), T2(ana2-mono), ..., T5, deta1, deta2, deat3, deat4, deta5, detsq
Further raw data treatment and summing of spectra
sqwadd is a tool to prepare IXS-spectra for further fitting after they have been extracted from the standard SPEC-data file (extraction done via newplot). It allows to display and inspect the individual spectra of each scan and each analyser, e.g. to compare repeated spectra, centre the individual spectra (by pre-fitting them), sum together several spectra (repetitions or extensions of spectra) and save the spectra or sums of spectra converted from a temperature-axis to an energy axis.
sqwadd has to be started in the directory, where you have saved the extracted file; the tool will save the results in this same directory (e.g. on brahe.esrf.fr: ~/experiments/HS2000.) The tool opens in a window offering pull-down menus as well as click-buttons for the tasks to do.
Load data files: menu FILE – OPEN and load the data to be treated together.
Choice of reflection order: Choose the correct incident photon energy by selecting the corresponding correct backscattering reflection ("Bragg Reflection", e.g. 999).
Output file names: Filenames have to be given for each analyser individually, either under "SCANS" or under "OUTPUT".
Selection of the analyser: The analyser whose spectrum shall be treated, has to be chosen by clicking on the corresponding button on the main panel.
The data-treatment is done in 3 steps. The described procedures have to be repeated for every spectrum and every analyser of interest. Note that this procedure is indispensable, even if you have only a single file. In order to fit the data with fitvie (see below) you need a file with a constant step in temperature (energy).
1. Determination of the elastic line:
The first step consists of determining the position of the elastic line, corresponding to zero energy-transfer. The fitting is started by clicking on the taskbar-button with two Gaussian-like profiles, which opens an additional window to first "estimate" the values of the fitting parameters and "start" the fitting itself. By default the fits are done with Lorentzian function plus some background, but other functions can be chosen from the menu. The number of peaks is "found" automatically and cannot be changed, even not by changing the type of fit-function. Note, that only the Lorentzian-fit results are taken into account automatically for the further treatment; if the fits are done using other fit-functions (Gaussian, PseudoVoigt etc.) the values have to be entered manually. If the number of peaks offered or the quality of of fit is not at all convenient, the zero-energy-transfer can also be estimated with help of crosshairs (to be found in the "GRAPH"-pull-down menu), and the value found can then be entered manually. At the end of the procedure, the column “SHIFT” in the sheet “SCANS” should contain the correct value by which the spectrum has to be shifted in order to have the elastic line centred at zero.
2. Centring of the spectrum:
The centring of the spectrum is done by clicking on the taskbar-button showing a profile with crosshairs (or by choosing the "CENTER"-item in the pull-down menu). The procedure consists to shift the spectrum by the chosen amount, and interpolate the spectrum in order to obtain an equally spaced grid on the x-axis. This is necessary, if the data shall be fitted with minuit.
3. Summing the spectra:
The third taskbar-button, displaying a profile with a "+"-symbol, then sums the highlighted spectra together, converts them into an energy (meV-) scale and saves the results under the filename given ("OUTPUT" or "Output File Name"). If you have prepared as described above the spectra for all analysers and have defined all filenames for the results, you can do the summing-up by only clicking on the "SUM ALL"-button, instead of doing the summing-up for each analyser individually.
The sqwadd-application window itself will not display whether or not it performed the tasks, but the window, where you started the tool will display comments on the tasks (for the centring, summing-up, saving into the file etc.).
To be able to use other software to continue to treat these summed-up data, here the description of the columns in the sqwadd-output (ASCII-) file:
1st col.: energy transfer 2nd col.: counts (normalised to the average monitor) 3rd col.: error bar of col. 2 4th col.: unknown purpose 5th col.: original counts (as summed up) 6th col.: summed-up monitor (from the ione-monitor in the SPEC-datafile) 7th col.: error bar of col. 5
On-line fitting with minuit
The fitting is done in your analysis directory (users/opid28/experiments/hsNNN). For fitting with the minuit library, called by the program fitvie (n excitations: Lorentzian line shape, damped harmonic oscillator or other line shapes), you need a file with a constant step in temperature (energy), so you have to use sqwadd beforehand, even for one single scan. You need the summed data and the actual resolution function files (ask the local contact), and to prepare an input file for each scan you want to analyse.
1. Preparation of the input file:
Copy the standard input file exvie.inp (for fitvie) on brahe in users/opid28/usermacros to your directory: users/opid28/experiments/hsnnnn. Copy the resolution file (named aNresJ or aNexpJ, N=1…5, J=8, 9, 11, with extensions .dat, .090 or .200, depending on the energy resolution file corresponding to your experiment) in the resolution directory users/opid28/resolutions/validresolutions to your directory: users/opid28/experiments/hsnnn. N is the analyser number, J the Si reflection order, “res” signifies resolution data (fitted or interpolated) centred around zero energy transfer and normalised to integral=1. “exp”-files are close to experimental data, non-normalised, non-interpolated or –fitted, only centred around zero energy transfer. Example: a3res11.200 denotes the experimental resolution function of analyser #3 at the silicon (11,11,11) reflection order valid for a 200mA-mode of the ESRF (e.g. uniform filling or 2/3-filling; not valid for e.g. 16bunch mode). The format of the input file is as follows:
| n005_RT.cen | ! data input file |
| n005_RT.fi2 | ! fitted output file |
| a2res9.200 | ! resolution file |
| DHO | ! fitting model |
| n005_RT.do2 | ! file do for c-plotting |
| n005_RT.pa2 | ! fitted parameters file |
| 294. | |
| 1. |
! 1/fraction of step used for convolution ! (higher=slower) |
| NbSe3; Q=(4, 0.05, 4); T=294K | |
| 100 | ! int. time |
| no |
! yes=viscoelastic otherwise off (if no, FIX SA, ta, aa, ! bb) |
| 1 | ! Number n of excitations (MAX 12) |
| dho | ! first excitation (dho, lor, gau, del) delete line if n=0 |
MINUIT : wathp
1 BG 0.5 0.01 0.001 20.0 2 SL 0.00 0.01 -10.0 10.0 3 ZR 0.00 0.01 -10.0 10. 4 GR 2.00 -.1 .000001 10. 5 PC 21.400 0.100 -1.0000 1000. 6 GC 0.0600 -.01 .000001 10.0 7 SA 2.00 -.01 -100.00 100. 8 ta 0.5000 -.01 .000001 10.0 9 aa 20.000 -.01 .000001 100. 10 bb 10.000 -.01 .000001 100. 11 P1 0.04 .001 .0001 100. 12 O1 1.20 .01 1.00 10.0 13 G1 0.10 -.01 .10 10.
| SET PRI | 2 |
| SET ERR | 1 |
| FIX | 2 |
| FIX | 3 |
| FIX | 5 |
| SIM | |
| HES | |
| MIG | |
| HES | |
| EXIT |
1. line: name of data file (input data)
2. line: name of output file, containing the experimental data and the fit
3. line: file of the experimental resolution function with which the model function is convoluted
4. line: part of title of the print-out describing the type of phonon line shape used:
| dho: Damped Harmonic Oscillator | |
| lor: Lorentzian functions | |
| gau: Gaussian function | |
| del: delta function (harmonic oscillator) |
5. line: name of the C-PLOT do-file with which one can visualise and print the results
6. line: name of the parameter file (output file with the calculated parameters)
7. line: temperature in degree Kelvin (real number)
8. line: comment (title for the do file)
9. line: integration time of each data point
10. line: visco(yes/no) if yes it add a viscoelastic line shape to the model ONLY for fitvie
10. line: number of excitations
11. line: name of the first excitation (dho, lor, del, gau...) next lines: name of the other excitations (if it is the case).
The meaning of the fitting parameters is as follows:
| BG | background |
| SL | linear slope of the background |
| ZR | energy position of the elastic line |
| GR | only used if one convolutes with a Lorentzian function, otherwise has to be kept fixed |
| PC | central line intensity |
| GC | central line width |
| SA | visco-elastic intensity (ONLY for fitvie) |
| TA | relaxation time (ONLY for fitvie) |
| aa |
|
| bb |
|
| P1 | 1st. phonon line intensity |
| O1 | 1st. phonon line energy |
| G1 | 1st. phonon line width |
| P2 | 2nd. phonon line intensity |
| O2 | 2nd. phonon line energy |
| G2 | 2nd. phonon line width |
.......and so on up to a maximum of 12 excitations.
GN for delta function is just a "take place" line, the value must be there, but it is just read, not used, otherwise if GN=0, the program takes automatically a delta line-shape, whatever you choose as excitation.
The parameters of the visco-elastic (v.e.) model must be there only if you run fitvie, if 'visco' is off, they are just "take place" lines.
The following lines are standard commands to execute the minuit libraries minimisation, the only ones you need to specify is the FIX line: put 1 FIX line for each parameters you want to minimise, with the number of the parameter (FIX 1 to block BG, FIX 3 to block ZR and so on).
The columns of the lines BG and following are:
1st: number of fit parameter
2nd: name of fit parameter (BG, etc.)
3rd: starting value of the fit parameter
4th: first step of change of the parameter during fitting
5th: lower limit of allowed range for the fit parameter
6th: upper limit of allowed range for the fit parameter
The 4th column offers another means to fix (block) a fit parameter: if a negative value is given, the parameter will be kept constant during the fit.
2. Running the fitting program:
> fitvie < name_file.inp (example: fitvie < ger12.inp)
3. Plotting the result of the fit:
The results of the fit can be visualised and printed using CPLOT with the CPLOT macro generated by fitvie (with the name you put on line 5 of the input file e.g. on CPLOT :
PLOT>do cpfilename.do)
To print, modify the CPLOT macro, putting the zi x11 statement in comment (#zi x11), and run the macro again.
To be able to use any other software to visualise the fit-curves, here the description of the columns in the fit-output (ASCII-) file (the filename is defined in the 2nd line of the input-file):
1st col.: energy transfer 2nd col.: data 3rd col.: error bar of data 4th col.: total fit (sum of all contributions) 5th col. (and following): fits of the various contributions to the spectrum fitted
e.g. elastic line, excitations
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.