- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2009
- Structure of materials
- Direct synthesis of cubic ZrMo2O8 followed by ultrafast in situ powder diffraction
Direct synthesis of cubic ZrMo2O8 followed by ultrafast in situ powder diffraction
The cubic AM2O8 family of materials (A = Zr, Hf; M = W, Mo) has attracted considerable interest due to the negative thermal expansion properties of its members. Cubic ZrW2O8 is the most famous of these materials and contracts isotropically when heated with a thermal expansion coefficient of –9 x 10-6 K-1 between 2 and 300 K. It has two potential drawbacks for any practical applications: a discontinuity in cell parameters around 177°C associated with an order-disorder transition, and a transition to a denser structure at pressures above ~0.2 GPa. In contrast, cubic ZrMo2O8 shows no such discontinuities in cell parameter and pressure-induced transitions are reversible at room temperature. The major issue is the difficulty of its synthesis. Cubic ZrMo2O8 was thought to be metastable at all temperatures (less interesting monoclinic and trigonal forms are thermodynamically stable) and has only been prepared by complicated low temperature routes via controlled decomposition of precursors. Recently we obtained tantalising evidence in the laboratory that it might be possible to prepare this metastable phase at high temperatures (~1177°C) in a restricted temperature-time window.
We therefore turned to rapid in situ experiments at ID11 to unravel the solid-state chemistry involved in this synthesis. In a typical experiment we pre-mixed metal oxide reactants in a thin-walled Pt capillary then transferred them into a pre-heated mirror furnace (Figure 44) and collected a series of powder diffraction patterns. Using a wavelength of 0.2 Å, we could penetrate the Pt capillary and record data of sufficient quality for Rietveld refinement with a time resolution of 0.1 s. This setup allowed synchronous monitoring of the metal oxides as they reacted at high temperatures.
 |
|
Fig. 44: Mirror furnace. |
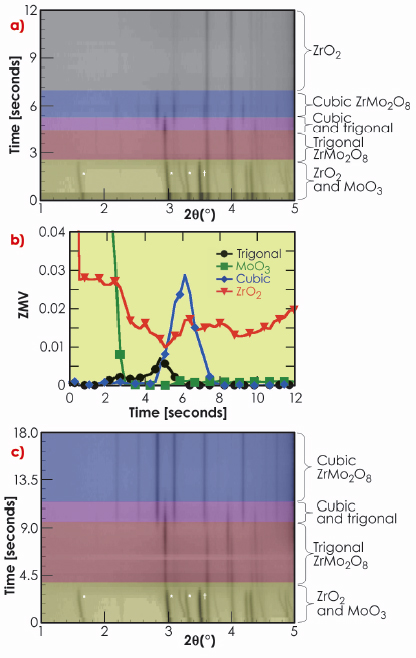
Figure 45 shows data from one of our first experiments on the ZrO2 + 2MoO3 reaction, which gave huge insight into the possible direct synthesis of cubic ZrMo2O8. Here the sample was introduced into the furnace preheated to 1250°C and the reagents reacted extremely rapidly. Figure 45a shows a two dimensional representation of the diffraction data and Figure 45b shows results from quantitative refinements with data plotted as “ZMV”, a measure of the relative amount of the phases present in the mixture. At low temperatures, the diffraction peaks correspond to ZrO2, MoO3 and the Pt of capillary walls as expected. Initially all peaks shift to lower 2q indicating positive thermal expansion on heating. Between approximately 1.6 and 2.5 s (T ~757°C and 987°C) peaks due to MoO3 disappear and those of trigonal ![]() -ZrMo2O8 are observed for the first time. A drop in the overall diffracted intensity occurs at this point, which is consistent with the melting of MoO3 (Tmelt = 795°C). Between 2.5 and 5 s (T ~1161°C) the amount of trigonal
-ZrMo2O8 are observed for the first time. A drop in the overall diffracted intensity occurs at this point, which is consistent with the melting of MoO3 (Tmelt = 795°C). Between 2.5 and 5 s (T ~1161°C) the amount of trigonal ![]() -ZrMo2O8 increases before gradually disappearing. New peaks corresponding to cubic
-ZrMo2O8 increases before gradually disappearing. New peaks corresponding to cubic ![]() -ZrMo2O8 appear from 4.5 s (T ~1127°C). These peaks disappear after a total elapsed time of 8 s (T ~1252°C) and ZrO2 is the only crystalline material remaining. On quenching after 120 s (T falls from ~1247°C to ~427°C within 8 seconds), significant quantities of cubic ZrMo2O8 were again observed in the diffraction data, suggesting a molten phase at the highest temperatures.
-ZrMo2O8 appear from 4.5 s (T ~1127°C). These peaks disappear after a total elapsed time of 8 s (T ~1252°C) and ZrO2 is the only crystalline material remaining. On quenching after 120 s (T falls from ~1247°C to ~427°C within 8 seconds), significant quantities of cubic ZrMo2O8 were again observed in the diffraction data, suggesting a molten phase at the highest temperatures.
 |
|
Fig. 45: Data obtained for two reactions: a) (ZrO2):2(MoO3) at 1247°C and b) resulting phase fractions; c) (ZrO2):3(MoO3) at 1127°C. ZrO2 and MoO3 peaks are indicated by |
This experiment gave us many of the clues needed to synthesise the cubic phase. By lowering the temperature to ~1197°C we were able to stabilise cubic ZrMo2O8 over a period of 60 s, though with ZrO2 present as a second phase. By using a small excess of MoO3 we were able to prepare the cubic phase as the only crystalline product (Figure 45c) in a period of just 9 s.
In summary, the high flux, high energy beam available at ID11 coupled with the fast data acquisition achievable with the FReLoN camera have given unique insight into the synthesis of cubic ZrMo2O8. We have shown that an apparently metastable material can be prepared directly from its component oxides and in a matter of seconds at high temperature. The extremely fast reaction rate seems to be due to the formation of molten MoO3 a couple of seconds into the experiment which acts as a reactive flux. Online (and almost real time!) data analysis was essential to allow us to optimise the reaction conditions during the experiment and enabled us to produce the pure target material. These methods could be readily applied to a host of other fascinating systems.
Principal publication and authors
J.E. Readman (a), S.E. Lister (a), L. Peters (b), J. Wright (c) and J.S.O. Evans (a), J. Am. Chem. Soc. 131, 17560 (2009).
(a) University of Durham (UK)
(b) Christian-Albrechts-Universität zu Kiel (Germany)
(c) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.