- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Dynamics and extreme conditions
- Water: mixture or continuum?
Water: mixture or continuum?
Water is one of the most well-studied substances, yet we still do not have a complete description of its structure. The complex hydrogen-bond network that gives rise to water’s remarkable properties is transient, requiring different descriptions at different time and length scales. Spectroscopic studies of water provide compelling but often conflicting information on the local structure and dynamics. One of the recurring questions that surfaces periodically is whether the liquid is best described as a continuum or a mixture at the molecular scale.
The earliest descriptions of water were mixture models, of miniature icebergs mixed with a “normal” liquid, which helped explain some of the anomalous properties of water like the density maximum at 4°C. Since the advent of molecular dynamics, simulations of varying complexity have regularly suggested a continuum model of liquid water. In this view, hydrogen bonds in the liquid are distorted, broken and reformed continuously, but the liquid itself is composed of a single component.
However, spectroscopic data can often be modelled by assuming the existence of distinct species with different local structures: a modern version of a mixture model with a tetrahedrally-coordinated ice-like component and a dense component with distorted or broken hydrogen bonds. While these mixture models are not supported by molecular dynamics simulations, they do have one very attractive feature: they establish a direct correspondence between the structure in the liquid phase and that of low-density and high-density amorphous ices, the existence of which is another peculiar and much discussed property of water. X-ray spectra in particular have recently been used to support the mixture model view of water structure [1]. Existing X-ray and neutron diffraction techniques are relatively insensitive to the details of hydrogen bonding in water. Therefore, further spectroscopic investigations are of interest in order to test the validity of the different models.
One particular spectroscopic result is often taken as direct evidence of a two-component structure: van’t Hoff behaviour, in which temperature-induced spectral changes can be viewed as changes in the populations between two components. Importantly, a van’t Hoff fit of temperature-dependent spectra gives a value for the enthalpy change in the conversion between the two components.
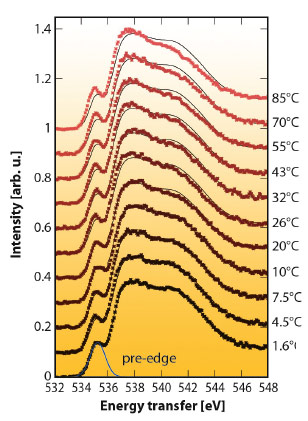
Using an in-house-developed temperature-controlled liquid flow setup at beamline ID16, we measured the oxygen near-edge spectrum of liquid water at various temperatures using X-ray Raman scattering. Thanks to excellent statistical accuracy and stability we can see a subtle and systematic evolution with temperature (Figure 49). We focused our attention on the pre-edge feature that has been discussed extensively in the literature [1]. In particular, it has been connected to distorted and broken hydrogen bonds, and has been used to support a two-component model.
 |
|
Fig. 49: The temperature dependence of the oxygen near-edge spectrum. A smoothed version of the 1.6°C spectrum is shown for reference throughout. |
We find that the temperature dependence of the pre-edge intensity in our data does indeed display van’t Hoff behaviour (Figure 50). However, the enthalpy difference, ΔH, between the two assumed components obtained from a van’t Hoff fit to our data is small, only 0.9 ± 0.2 kcal/mol. This is of the order of the latent heat of melting from ice Ih to liquid (1.43 kcal/mol), but much smaller than the strength of one hydrogen bond (out of two per molecule) in ice, 5.5 kcal/mol. In terms of hydrogen bond strengths, the two populations are found to be very similar. In our earlier work we showed that the near-edge region in various ice polymorphs is sensitive to non-hydrogen-bonded neighbours [2]. The current results indicate that the pre-edge in liquid water is sensitive to distortions that leave hydrogen bonds intact. Earlier studies proposing that severe distortion and breaking of hydrogen bonds is needed to produce the pre-edge feature should be revisited. Our results are furthermore consistent with the continuum models of molecular dynamics simulations, bridging an important gap between computer simulation and X-ray spectroscopy experiments.
 |
|
Fig. 50: The intensity of the pre-edge component displays van’t Hoff behaviour. A fit to the data gives an enthalpy change DH in the conversion between two components. |
Principal publication and authors
T. Pylkkänen (a,b), A. Sakko (a), M. Hakala (a), K. Hämäläinen (a), G. Monaco (b) and S. Huotari (a,b), J. Phys. Chem. B 115, 14544-14550 (2011).
(a) Department of Physics, University of Helsinki (Finland)
(b) ESRF
References
[1] For a recent review, see A. Nilsson and L. G. M. Pettersson, Chem. Phys. 389, 1-34 (2011).
[2] T. Pylkkänen, V. M. Giordano, J.-C. Chervin, A. Sakko, M. Hakala, J. A. Soininen, K. Hämäläinen, G. Monaco and S. Huotari, J. Phys. Chem. B 114, 3804-3808 (2010).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.