- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2007
- Structural Biology
- A bacterial key to host cell entry: Activation of the human receptor tyrosine kinase Met by a bacterial ligand
A bacterial key to host cell entry: Activation of the human receptor tyrosine kinase Met by a bacterial ligand
Listeria monocytogenes, a gram-positive, facultative intracellular human pathogen, employs the invasion protein InlB to enter normally non-phagocytic cells. This bacterial surface protein InlB binds to and activates the receptor tyrosine kinase Met on host cells [1]. Met activation leads to rearrangements of the actin cytoskeleton and ultimately causes uptake of L. monocytogenes. InlB is a multi-domain protein, but only its N-terminal half is essential for Met binding and activation. A capped leucine-rich repeat (Cap-LRR) domain of InlB harbours the high affinity Met-binding site and amino acids involved in InlB-Met interactions have been mapped to the concave face of the curved Cap-LRR [2]. Despite this high-affinity binding site, the Cap-LRR fragment of InlB alone cannot activate Met. Instead, activation additionally requires the so-called inter-repeat (IR) region, an immunoglobulin (Ig) like moiety that follows the Cap-LRR domain.
The extracellular domain of Met comprises a large seven-bladed ß-propeller (Sema domain), a small cysteine rich domain (PSI) and four immunoglobulin-like domains (Ig1 – Ig4). Of these six domains the Sema domain exclusively binds the physiological Met ligand hepatocyte growth factor (HGF). Although a crystal structure of the Met Sema and PSI domains in complex with one domain of HGF has been solved [3], the mechanism of Met activation by HGF remains elusive. HGF and InlB can bind Met simultaneously [1], but the InlB-binding site on the ectodomain of Met was unknown. Likewise the structural basis of InlB mediated Met activation and the function of the IR region of InlB were unclear.
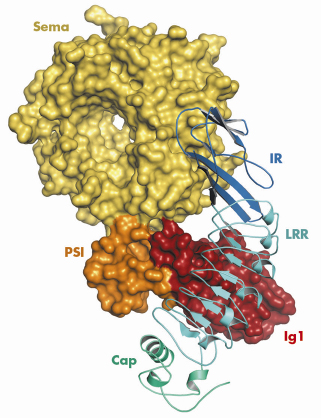
We determined the crystal structure of a complex between the Met-binding domain of InlB and a large fragment of the Met ectodomain using data collected on beamline ID23-2 (Figure 65). The kidney shaped InlB binds via the concave face of its Cap-LRR domain to the first immunoglobulin-like (Ig1) domain of Met. An unusual beta-hairpin extending from the core of Ig1 is pivotal for the interaction with InlB. This interface represents the primary contact between InlB and Met and involves five aromatic amino acid side chains that are exposed on the surface of InlB. The observed mode of binding is consistent with all biochemical data for InlB [1,2], but strikingly different from that of the physiological Met ligand HGF (Figure 66).
 |
|
Fig. 65: Structure of the InlB/Met complex. |
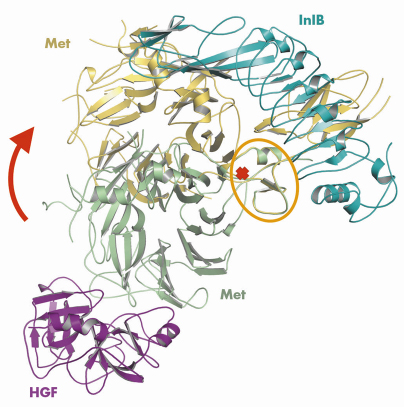
The structure additionally reveals an unexpected second region of contact between InlB and Met. This less prominent interface is formed between the InlB IR region and the Sema domain of Met (Figure 65). Binding studies had not predicted this contact, as it does not contribute measurably to binding affinity. However, it is essential for receptor activation. Comparison of our structure with that of HGF in complex with the Sema and PSI domains of Met reveals a dramatic conformational change in the Met ectodomain. The Met Sema domain undergoes a rigid body rotation of some 60° relative to its neighbouring PSI domain (Figure 66).
 |
|
Fig. 66: Overlay of structures of the InlB/Met and the HGF/Met complexes. The alignment was performed on the PSI domain (marked by the orange oval). |
The arrangement of the Met domains in the complex with InlB is governed by the two-site interaction between both proteins and, therefore, represents a uniquely defined conformation. This assumption is confirmed by a second structure of the complex that we obtained in a different crystal form (data also collected on beamline ID23-2). The overall domain arrangement is very similar in both structures of the InlB/Met complex and the second interface between the InlB IR and the Met Sema domain is preserved. We, thus, propose that the rigid InlB acts as a “molecular clamp” and locks the otherwise flexible Met receptor into a defined, signalling competent conformation. Whether and how this InlB induced conformation results in the expected dimerisation of the Met ectodomain needs to be addressed by future investigations.
References
[1] Y. Shen, M. Naujokas, M. Park, and K. Ireton, Cell 103, 501–510 (2000).
[2] M.P. Machner, S. Frese, W.D. Schubert, V. Orian-Rousseau, E. Gherardi, J. Wehland, H.H. Niemann, and D.W. Heinz, Mol. Microbiol. 48, 1525–1536 (2003).
[3] J. Stamos, R.A. Lazarus, X. Yao, D. Kirchhofer, C. and Wiesmann, EMBO J. 23, 2325–2335 (2004).
Principal publication and authors
H.H. Niemann (a), V. Jäger (a), P.J.G. Butler (b), J. van den Heuvel (a), S. Schmidt (a), D. Ferraris (a), E. Gherardi (b) and D.W. Heinz (a), Cell 130, 235-246 (2007).
(a) Division of Structural Biology, Helmholtz-Centre for Infection Research, Braunschweig (Germany)
(b) MRC Centre and Laboratory of Molecular Biology, Cambridge (UK)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.