- Home
- News
- Spotlight on Science
- Crystal structure...
Crystal structure of poxvirus DNA polymerase
31-01-2018
Despite the eradication of smallpox virus in 1980, orthopoxviruses, which circulate widely in animals, remain dangerous pathogens. The crystal structure of the DNA polymerase of the prototype vaccinia virus is reported here. This protein is essential for the replication of the viral genome. The structure allows an understanding of anti-viral resistance and provides information about the unique mode of processivity factor binding.
One of the great success stories of modern medicine is the eradication of smallpox virus that was declared in 1980 after a long vaccination campaign with vaccinia virus. A risk remains today of the resurgence of smallpox virus from a frozen source such as the melting of permafrost [1]. Another risk is the spread of an orthopoxvirus from an animal reservoir to the human population. The present work focuses on vaccinia virus, which is a safe model system to study poxviruses. The high-resolution structure that has been obtained of components of the vaccinia virus DNA replication machinery will facilitate the development of drugs and help to understand orthopoxvirus drug resistance.
The crystal structure determination of vaccinia virus polymerase was challenging due to the radiation sensitivity of the long needle-like DNA polymerase crystals obtained from low amounts of protein produced in insect cells. The use of the helical scan capability at the ESRF with a simultaneous rotation and translation of the crystal during data collection together with wavelength optimisation at ID23-1 was essential (Figure 1). An experimental phasing (MIRAS) strategy based on the expected binding of gadolinium and lead cations by carboxylic acid clusters present in polymerase and exonuclease active sites of the polymerase was chosen and led to the 2.75 Å resolution structure. Additional small-angle X-ray scattering (SAXS) experiments carried out on BM29 were essential to validate a model of the DNA bound form of the polymerase required for the understanding of resistance mutations (Figure 2) and for the localisation of the processivity factor binding site.
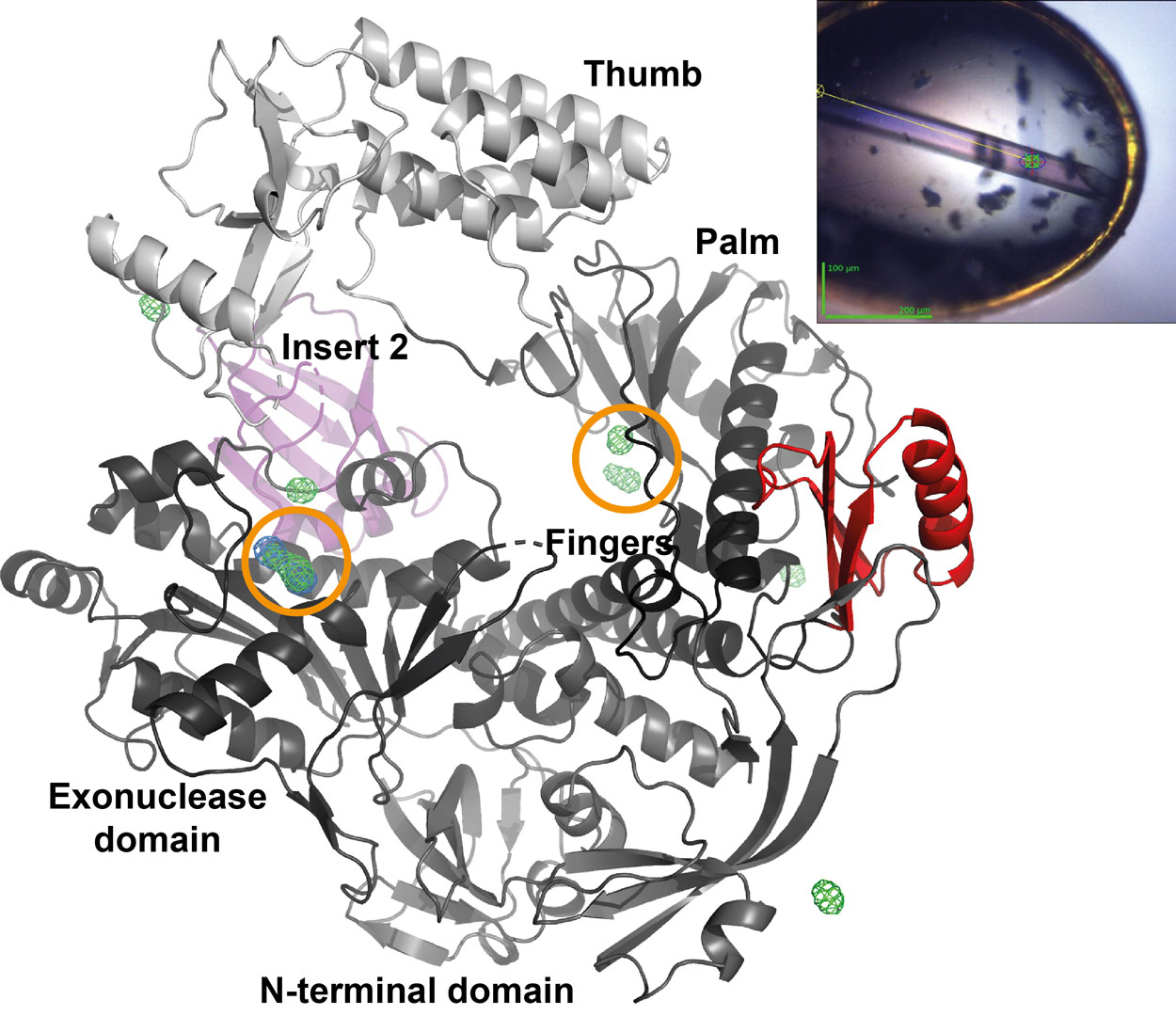 |
|
Figure 1. Overall structure of vaccinia virus DNA polymerase showing the canonical domains of family B polymerases. The poxvirus-specific insert 2 is shown in pink, insert 3 in red. Active sites are indicated by orange circles. Anomalous difference maps contoured at 10 σ show the position of Gd3+ ions (green) and Pb2+ ions (blue) used for experimental phasing. The insert shows a frozen crystal on the mini-diffractometer and the line along which the crystal was translated during the helical scan. |
The structure shows that the poxvirus enzyme is a family B polymerase close to eukaryotic polymerase δ [2] but with specific insertions, in particular insert 2 with unknown function and insert 3 at the periphery of the molecule (Figure 1). The binding site of the poxvirus specific processivity factor, which is formed by a heterodimeric protein complex [3], remained to be identified. For this purpose, the processivity factor domain interacting with the DNA polymerase was isolated using the recombinant ESPRIT [4] technology for the identification of soluble protein fragments. The obtained polypeptide was shown by SAXS to fold as a compact domain. As it was not possible to obtain a crystal structure of the complex, a biochemical approach using protection from deuterium-hydrogen exchange coupled to mass spectrometry was chosen. The interacting residues of the polymerase were identified as part of insert 3 and were confirmed by site-directed mutagenesis. The interaction of the DNA polymerase with its processivity factor through an insert in the palm domain is a novel observation since family B polymerases usually interact with ring-shaped processivity factors via their extreme C-termini.
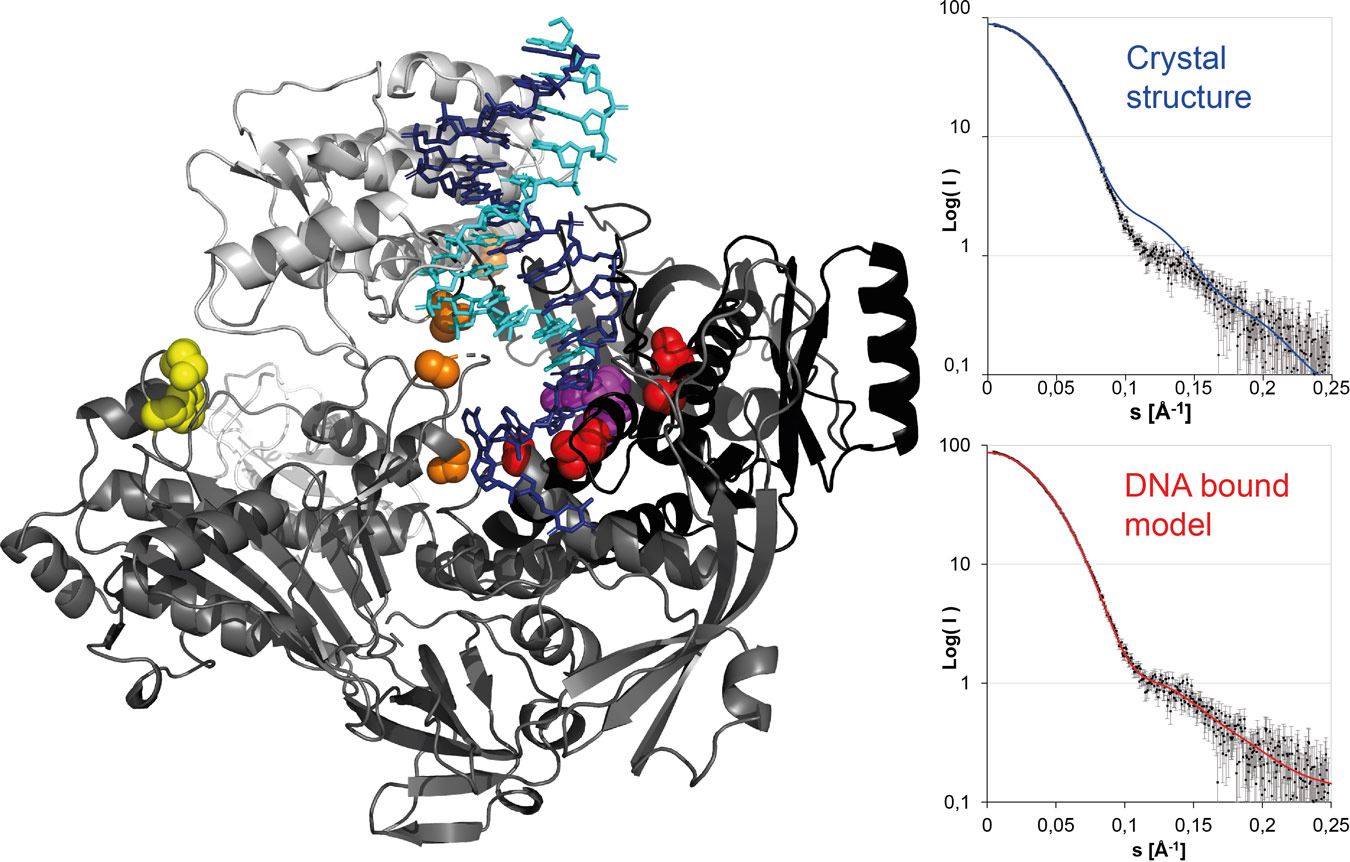 |
|
Figure 2. (Left panel) Model of the vaccinia virus DNA polymerase in elongation mode with bound template strand (blue), the newly synthesised strand (cyan) and the incoming nucleotide in space-filling representation (magenta). Positions of resistance mutations, shown as coloured spheres, affect the elongation site (red), the exonuclease site (yellow) or DNA switching between the two sites (orange). The domain movements in the model, compared to the crystallised DNA-free form, were confirmed using SAXS (right panel, calculated curve from the DNA-free model in blue, from the DNA-bound model in red, experimental points from the vaccinia virus polymerase/DNA complex in black). |
SAXS studies indicated that the DNA polymerase structure closes upon binding to a template dsDNA in the same way as other family B polymerases. A model of the DNA-polymerase complex (Figure 2) was obtained that permitted an understanding of resistance mutations against polymerase inhibitors; for example, the molecule cidofovir that is used in the treatment of poxvirus infections. Resistance mutations affect either the polymerase active site, the exonuclease active site used in proof-reading (which excises chain-terminating inhibitors such as cidofovir) or the switching between both activities.
Principal publication and authors
The vaccinia virus DNA polymerase structure provides insights into the mode of processivity factor binding, N. Tarbouriech (a), C. Ducournau (b), S. Hutin (a), P.J. Mas (c), P. Man (d, e), E. Forest (a), D.J. Hart (a, c), C.N. Peyrefitte (b, f), W.P. Burmeister (a), F. Iseni (b), Nature Communications 8, 1455 (2017); doi: 10.1038/s41467-017-01542-z.
(a) Institut de Biologie Structurale, CEA, CNRS, Université Grenoble Alpes, Grenoble (France)
(b) Unité de Virologie, Institut de Recherche Biomédicale des Armées, Brétigny-sur-Orge (France)
(c) Integrated Structural Biology Grenoble (ISBG) CNRS, CEA, Université Grenoble Alpes, EMBL, Grenoble (France)
(d) BioCeV-Institute of Microbiology, Czech Academy of Sciences, Vestec (Czech Republic)
(e) Faculty of Science, Charles University, Prague (Czech Republic)
(f) Emerging Pathogens Laboratory, Fondation Mérieux, Lyon (France)
References
[1] P. Biagini et al., N Engl J Med 367, 2057–2059 (2012).
[2] M.K. Swan et al., Nat Struct Mol Biol 16, 979–986 (2009).
[3] C. Sèle et al., J Virol 87, 1679–1689 (2013).
[4] H. Yumerefendi et al., J Struct Biol 172, 66–74 (2010).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.