- Home
- News
- Spotlight on Science
- Redox-active MOF...
Redox-active MOF for reversible capture and release of halogens
10-05-2017
Chlorine and bromine are highly toxic and corrosive. A new material has been discovered that could make their storage safer and it has been characterised at the ESRF by X-ray absorption spectroscopy. This material is the first metal-organic framework able to reversibly capture and release halogens.
With an annual production of ca. 56 million tons, Cl2 is widely used in water sanitation and in the production of plastics, such as polyvinyl chloride and polyurethanes, as well as solvents and pesticides. Although used on a smaller scale, Br2 is a key element in flame retardants, agricultural products, and pharmaceuticals. The extreme toxicity, corrosiveness, and volatility pose serious challenges for the safe storage and transportation of these halogens, which play a key role in the chemical industry. Solid materials capable of forming stable nonvolatile compounds upon reaction with elemental halogens may partially mitigate these issues by allowing safe halogen release on demand. Here, we demonstrate that elemental halogens quantitatively oxidise coordinatively unsaturated Co(II) ions in a robust azolate metal−organic framework (MOF), Co2Cl2BTDD (1) (BTDD = bis(1H-1,2,3-triazolo[4,5-b],[4,5-i])dibenzo[1,4]dioxin), to produce stable and safe-to-handle Co(III) materials, namely Co2Cl4BTDD (2) and Co2Cl2Br2BTDD (3), featuring terminal Co(III)−halogen bonds.
 |
|
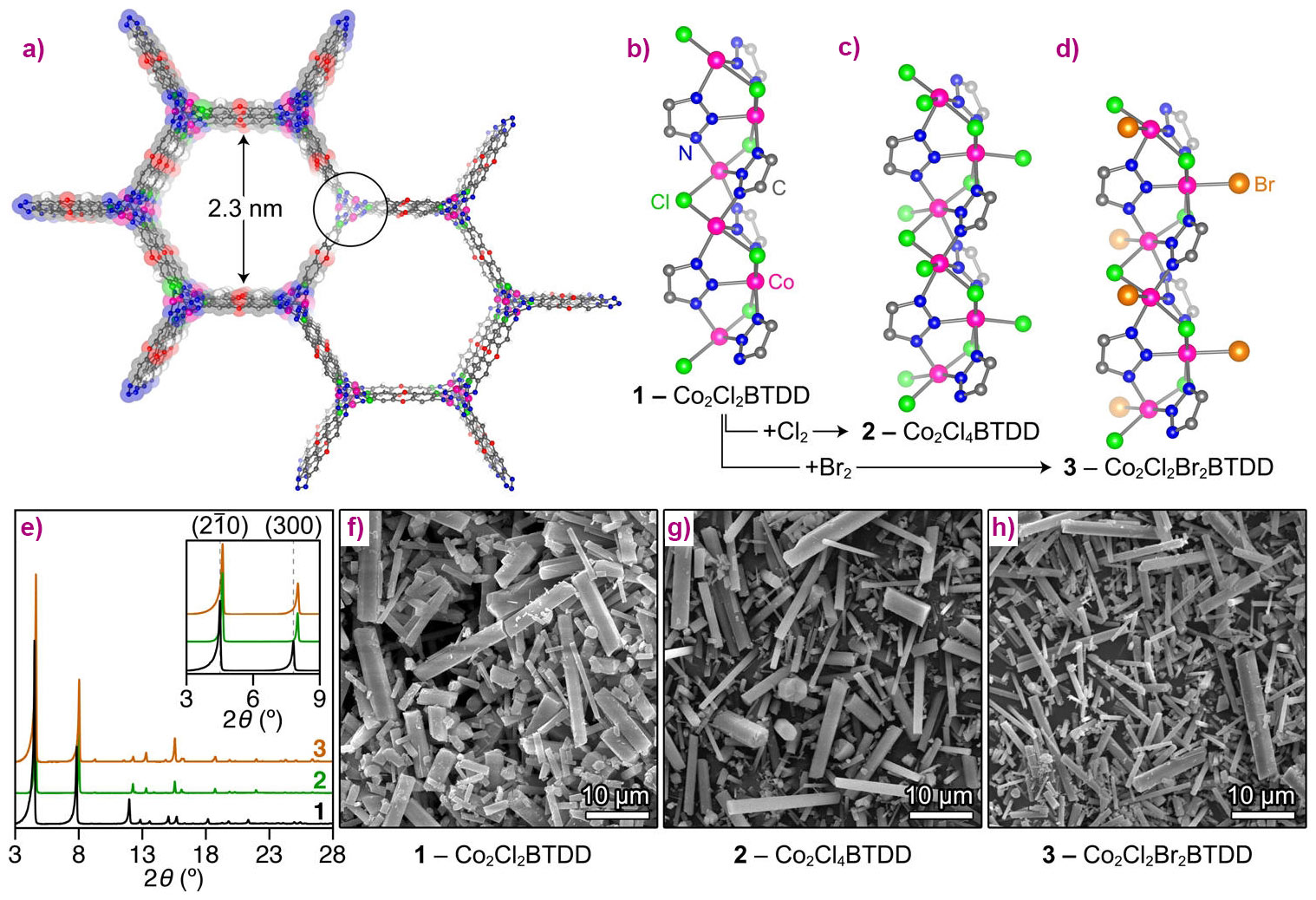
Figure 1. Structural and microscopy data. a) A portion of the structure of the parent Co(II) MOF Co2Cl2BTDD (1) projected along the c axis. b−d) Secondary building unit (SBU) structures and local coordination environments of the Co centres in (1), (2), and (3), respectively, as determined by NPD. e) Powder X-ray diffraction (PXRD) data for (1), (2), and (3) showing retention of crystallinity and the lattice contraction while moving from (1) to (2) and (3), as evidenced by the upshift in the 2θ values for the (2-10) and (300) reflections. F-h) Scanning electron micrographs of microcrystalline samples of (1), (2), and (3), respectively.. |
The parent material (1) was prepared according to a previously described method [1]. Its crystal structure was confirmed by neutron powder diffraction (NPD), revealing infinite −(Co−Cl)n− chains coiled into threefold spirals and interconnected by the bis(triazolate) linkers, resulting in a honeycomb-like structure with one-dimensional channels (Figure 1a). Reaction with gaseous Cl2 or Br2 led to rapid oxidation of Co centres of the material from Co (II) to Co(III) due to the coordination of chloride or bromide ligand, resulting in the formation of (2) or (3), respectively (Figure 1 b-d). The stoichiometry of the oxidation was confirmed by microelemental analysis and magnetometry, while retention of the crystallite shape and morphology was checked by SEM (Figure 1 f-h). Consequent structural changes had noticeable but rather small effects on the XRD pattern, since the long-range order of the material was not significantly perturbed (Figure 1e). However, the short-range local environment of Co centres was considerably altered, which made the subsequent element-selective X-ray absorption spectroscopy (XAS) measurements, performed at beamline BM23, very informative. First, ex situ XANES spectra of the parent material (1) and its oxidised counterpart (2) exhibited pronounced chemical shift of the Co K-edge, unambiguously confirming the 2+ and 3+ oxidation state of Co centres before and after treatment, respectively (Figure 2a). EXAFS data indicated a higher coordination number for the Co-Cl shell in (2), which is direct evidence that Co centres gain one Cl extra-ligand in addition to the two Cl neighbours that are part of the framework (Figure 2 b,c).
 |
|
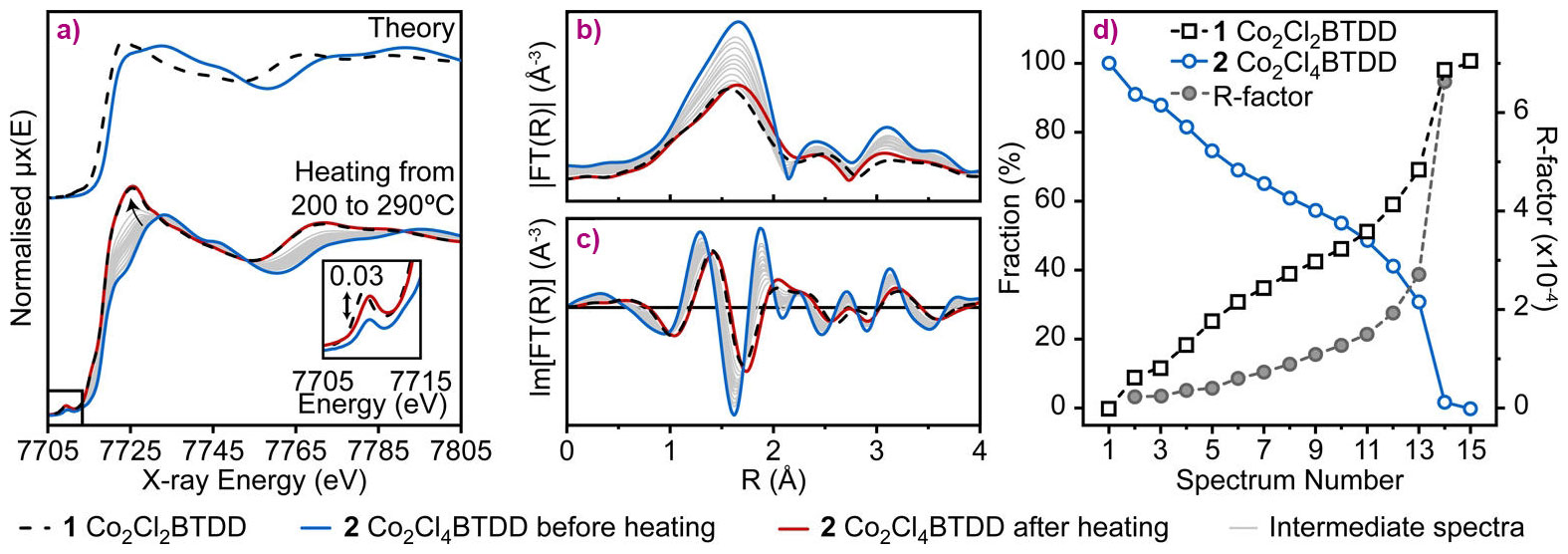
Figure 2. Co K-edge XAS characterisation of (1) and (2). (A) (top) Theoretical XANES spectra for (1) and (2) calculated from the NPD structures. (bottom) Experimental XANES spectra at room temperature for (1), (2), (2) after heating, and intermediate states collected in situ in the 200−290°C range during the thermal treatment of (2); (B) Magnitudes and (C) imaginary parts of the phase-uncorrected Fourier transformed k2-weighted EXAFS spectra; (D) Linear combination analysis of the whole series of in situ XANES spectra using the spectra of (1) and (2) at room temperature as references: relative fraction (open symbols) and fit quality factor (solid symbols). |
The reversibility of the Cl2 adsorption process was demonstrated by in situ XAS measurements performed upon heating (2) to 290°C (Figure 2 a-c). EXAFS data show a gradual decrease of the coordination number for the Co-Cl shell, while XANES spectra evidence reduction of Co centres. Importantly, both EXAFS and XANES spectra of (2) at the final stage of thermal treatment are very close to those of the parent material (1), proving the reversibility of Cl2 uptake on the local scale. Additionally, Cl2 release was quantified by linear combination analysis of the in situ XANES spectra, using the spectra of (1) and (2) at room temperature as references. It indicated almost complete conversion of Co (III) sites back to Co (II), but the elevated R-factor at the final stages of treatment suggested the formation of a minor fraction of unreacted Co in a slightly distorted environment, which explains the mismatch with the thermal gravimetric analysis (TGA) results, which indicate ca. 80% Cl release. Based on experimental evidence and subsequent density functional theory (DFT) calculations, a probabilistic model of Cl release by (2) was constructed. With the only initial criterion being pairwise vicinal removal of halogens, this model predicts a residual Co(III)−Cl content of 13.5%, corresponding to halogen release of 86.5% of the theoretical maximum.
These results demonstrate the ability of a Co(II) azolate MOF to uptake and release elemental halogens reversibly, and pave the way to the design of other porous materials geared toward the capture and storage of toxic, corrosive gases through reversible chemisorptive mechanisms.
Principal publication and authors
Reversible capture and release of Cl2 and Br2 with a redox-active metal−organic framework, Y. Tulchinsky (a), C.H. Hendon (a), K.A. Lomachenko (b,c), E. Borfecchia (d), B.C. Melot (e), M.R. Hudson (f), J.D. Tarver (f,g), M.D. Korzyński (a), A.W. Stubbs (a), J.J. Kagan (h), C. Lamberti (c,d), C.M. Brown (f), and M. Dincă (a), J. Am. Chem. Soc., 139, 5992-5997 (2017); doi: 10.1021/jacs.7b02161.
(a) Department of Chemistry, Massachusetts Institute of Technology, Cambridge (USA)
(b) ESRF
(c) IRC “Smart Materials”, Southern Federal University, Rostov-on-Don (Russia)
(d) Department of Chemistry, NIS, CrisDi, and INSTM Centre of Reference, University of Turin (Italy)
(e) Department of Chemistry, University of Southern California, Los Angeles (USA)
(f) Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg (USA)
(g) National Renewable Energy Laboratory, Golden (USA)
(h) Department of Mathematics, Weizmann Institute of Science, Rehovot (Israel)
References
[1] A.J. Rieth et al., J. Am. Chem. Soc. 138, 9401 (2016).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.