- Home
- News
- Spotlight on Science
- Structure and catalytic...
Structure and catalytic regulation of P. falciparum IMP-specific-nucleotidase revealed
26-01-2021
X-ray crystallography studies at the ESRF combined with comprehensive substrate screening reveal that the IMP-specific-nucleotidase 1 in the malaria-causing parasite Plasmodium falciparum is allosterically activated by ATP, and that complex rearrangements of domain organisation are tightly associated with catalysis. These results may contribute to designing possible malaria transmission-blocking agents.
P. falciparum (Pf), the parasite which causes the most severe form of malaria, requires mosquitoes and humans to complete its complex life cycle, with transmission of the disease mediated by the bite of an infected female Anopheles mosquito. In order to survive, the parasite makes use of a purine salvage pathway, and salvages nucleobases and nucleosides directly from the host in order to ensure nucleotide availability for its development. Hence, enzymes involved in this pathway constitute important targets for the development of new antimalarial drugs [1,2].
Indeed, while residing inside the red blood cells, adenosine and hypoxanthine salvaged from the human host are converted into inosine monophosphate (IMP), which is a precursor for the synthesis of both adenosine monophosphate (AMP) and guanosine monophosphate (GMP), providing indispensable nucleotides to the parasite [3]. In order to contribute to the understanding of the functioning of enzymes from this essential metabolic pathway, the IMP-specific nucleotidase, PfISN1 was studied.
IMP-specific 5’-nucleotidases (ISN1, EC 3.1.3.99) catalyse the de-phosphorylation of ribo- and deoxyribonucleoside monophosphates to their corresponding nucleosides, and possess the four characteristic Haloacid Dehalogenase (HAD) superfamily motifs. Although this family of nucleotidase is present only in eukaryotes, and largely confined to fungi, interestingly, in Plasmodium species, the ISN1 gene is restricted to only those parasites infecting humans, primates and birds. Indirect immunofluorescence microscopy using anti-PfISN1 antibodies showed cytoplasmic localisation in the intra-erythrocytic asexual and sexual stages of P. falciparum, an observation further confirmed by live-cell imaging.
In order to elucidate the relationship between the three-dimensional structure and the biochemical and physiological functions of PfISN1, its substrate specificity and kinetic parameters were determined. Data collection and testing of the diffraction power of a very high number of crystals using ESRF beamlines ID23-1, ID23-2, ID29, ID30A-1, ID30A-3 and ID30B were essential to the determination of crystal structures. Using data collected on ID29, the first crystal structure of PfISN1 (and of an ISN1-type nucleotidase) was solved from X-ray diffraction data on a selenomethionine derivative crystal (crystallised in the presence of ATP) by SAD phasing. All the remaining crystal structures of PfISN1, comprising the apo-enzyme, variants and complexes with various ligands adopting both ‘open’ and ‘closed’ conformations, and forming the basis for the mechanistic understanding of the enzyme, were solved by molecular replacement, using the ATP-bound structure as a search model. Variants and complexes include an inactive mutant D172N (full-length and ∆N30-truncated forms) bound to IMP and Mg2+, ∆C10-truncated, ∆N59-truncated and ATP-bound forms.
In the crystals, and despite a number of conformational variations of the solved structures, all adopt a tetrameric assembly displaying a cross-like shape (Figure 1). This is in agreement with the quaternary solution structure determined using small-angle X-ray scattering (SAXS), for which data were collected on beamline BM29, and negative staining electron microscopy data collected at the IBS electron microscope facility. Each subunit consists of four domains: an ‘N-Terminal Regulatory Domain’, an ‘Oligomerisation Domain’, a HAD-like Catalytic Domain and a ‘C2-type’ cap domain (Figure 1).
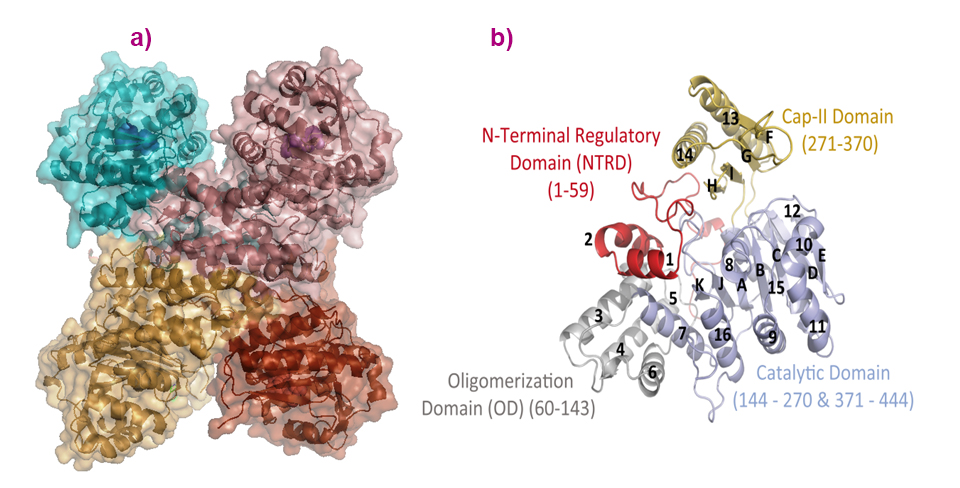
Fig. 1: a) The quaternary structure of PfISN1 is a tetramer, and (b) monomers are formed by the catalytic domain (light blue), oligomerisation domain (grey), cap-domain (sand) and N-terminal regulatory domain (red).
Structural and functional studies revealed the complex molecular reaction mechanism employed in this novel family of 5’-nucleotidases. An essential observation, amongst others, was that this enzyme is allosterically activated by ATP, which binds to a cleft formed by helices from the oligomerisation domain and from the catalytic domain, and which turns out to be at the very place in which the C-terminal (loop) is situated in the apo-form (Figure 2). Besides identifying amino acid residues critical for IMP binding and catalysis, this work also sheds light on how regulation is carried out via inter-domain and inter-molecular interactions. Overall, these results make it possible to suggest a detailed reaction mechanism carried out in four steps, implying important conformational changes and highlighting essential amino acid residues.
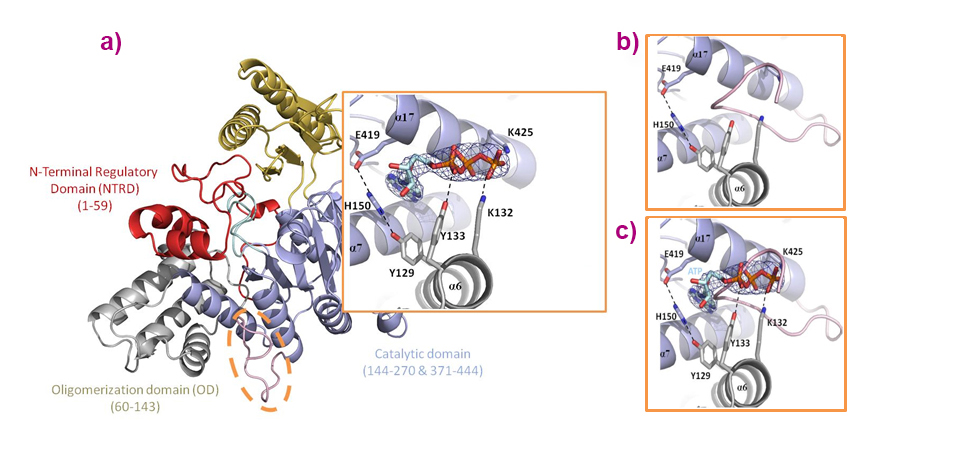
Fig. 2: a) Overall structure of the PfISN1-ATP structure with a close-up of the ATP binding site, highlighting that (b) ATP (allosteric activator) resides in a cleft formed by helices originating from the oligomerisation domain (grey) and from the catalytic domain (light blue). c) ATP binds at the very place where the C-terminal loop (L430-Q444) is situated in the apo-form.
The fact that the ISN1 gene is upregulated in female gametocytes suggests its role in this developmental form of the parasite that transitions upon fertilisation to ookinetes, zygotes and lastly sporozoites – the form that is eventually transmitted by mosquitoes. Inhibitors of PfISN1, developed through structure-based drug design, have the potential to interfere with female gametocyte development, and thereby serve as transmission blocking agents.
Principal publication and authors
Structure and catalytic regulation of Plasmodium falciparum IMP specific nucleotidase, L. Carrique (a), L. Ballut (a), A. Shukla (b), N. Varma (b), R. Ravi (b), S. Violot (a), B. Srinivasan (b), U.T. Ganeshappa (b), S. Kulkarni (b), H. Balaram (b), N. Aghajari (a), Nat. Commun. 11(1), 3228 (2020); doi: 10.1038/s41467-020-17013-x.
(a) Molecular Microbiology and Structural Biochemistry – CNRS/University of Lyon 1, Lyon (France)
(b) Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore (India)
References
[1] R.G. Ducati et al., Future Med. Chem. 5, 1341-1360 (2013).
[2] L. Ballut et al., Nat. Commun. 6, 8930 (2015).
[3] M.B. Cassera et al., Curr. Top. Med Chem. 11, 2103-2115 (2011).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.