- Home
- News
- Spotlight on Science
- The structure of...
The structure of an activated G protein coupled receptor bound to an engineered G protein
29-11-2016
Development of an engineered G protein, mini-Gs, has simplified the structure determination of a mini-Gs—bound G protein-coupled receptor (GPCRs) in a fully-active state. Similar strategies can now be applied to other GPCRs, which will advance our understanding of GPCR mechanism and facilitate structure-based drug design.
G protein-coupled receptors (GPCRs) are integral membrane proteins that are found at the surface of every cell in the human body. They are essential constituents of the molecular signalling system that choreographs the interplay between all our internal organs so that we can function as a human being. There are about 750 different GPCRs encoded by the human genome and they are responsible for all aspects of physiology involving intercellular communication. The adenosine A2A receptor (A2AR) is activated by adenosine, a cytoprotective modulator that is released during the stress response of an organ or tissue. The receptor is inhibited by drugs such as caffeine and is a potential target for the treatment of Parkinson’s disease, attention deficit hyperactivity disorder (ADHD) and in combination therapies for cancer.
Over the past 10 years there has been an explosion in our understanding of the molecular details of the structure of GPCRs. The structures have facilitated an in-depth understanding of how an agonist, such as adrenaline, binds to a receptor and alters its structure to facilitate the formation of an activated state that can, in turn, bind to a G protein. The G protein is then itself activated and can directly interact with other proteins that increase or decrease the concentrations of intracellular second messengers such as cAMP and Ca2+ that alter cellular function. To understand in structural terms how a receptor works we therefore need to have at least two structures of any given GPCR, a structure of the receptor in the inactive state bound to an antagonist and a structure in the activated state bound to a G protein. Structure determination of GPCRs in the inactive state is challenging, but is possible due to the development of many protein engineering strategies that stabilise receptors during crystallisation. The inactive state of a GPCR is invariably more stable than the activated state where the intracellular surface of the receptor opens up to allow the binding of a G protein. Thus the activated states of GPCRs are much more difficult to determine. This is highlighted by the publication of only a single structure of a GPCR-G protein complex, β2-adrenergic receptor (β2AR) bound to Gs, which contributed to the winning of a Nobel Prize by Brian Kobilka in 2012. If it is so difficult to crystallise a GPCR-G protein complex, how are we to determine structures of all the other GPCRs in a fully active state? In the absence of these structures it will also be difficult to understand the specificity of G proteins for GPCRs and, in turn, how hormones regulate effects in a cell that contain multiple different receptors that can each couple to multiple different G proteins.
Our previous work demonstrated that thermostabilising a GPCR can facilitate its structure determination, regardless of the affinity of the ligand bound to the receptor and whether this is an agonist or antagonist. This has also opened the door for the first time to structure-based drug design applied to GPCRs. However, in order to crystallise the active state of a receptor it must be bound to a G protein or a functional mimetic such as a nanobody. Heterotrimeric G proteins are large (90 kDa) multi-subunit proteins that are both unstable and conformationally dynamic when coupled to a GPCR. We therefore developed a novel minimal G protein, mini-Gs, in which the single G protein domain that mediates virtually all the contacts with the GPCR was isolated and thermostabilised [1]. Mini-Gs is only 28 kDa in size and recapitulates all the pharmacological effects of a heterotrimeric G protein binding to a GPCR. However, because mini-Gs is small and thermostable, the complex it formed with A2AR was amenable to crystallisation in a short chain detergent, octylthioglucoside, by vapour diffusion. Following data collection at beamline ID23-2, the structure (Figure 1) was determined to 3.4 Å resolution and provided the molecular details of how mini-Gs binds to A2AR and also a clear view of the ligand binding pocket in the activated state. The latter was surprising as it appeared virtually identical to a previously determined structure in the absence of bound G protein, thus suggesting that the energy landscape of different receptors such as A2AR and β2AR are very different. The molecular details of how mini-Gs binds to A2AR also differ in some aspects to how Gs binds to β2AR, despite the overall topologies being virtually identical. This is to be expected given the different amino acid sequences between the two receptors.
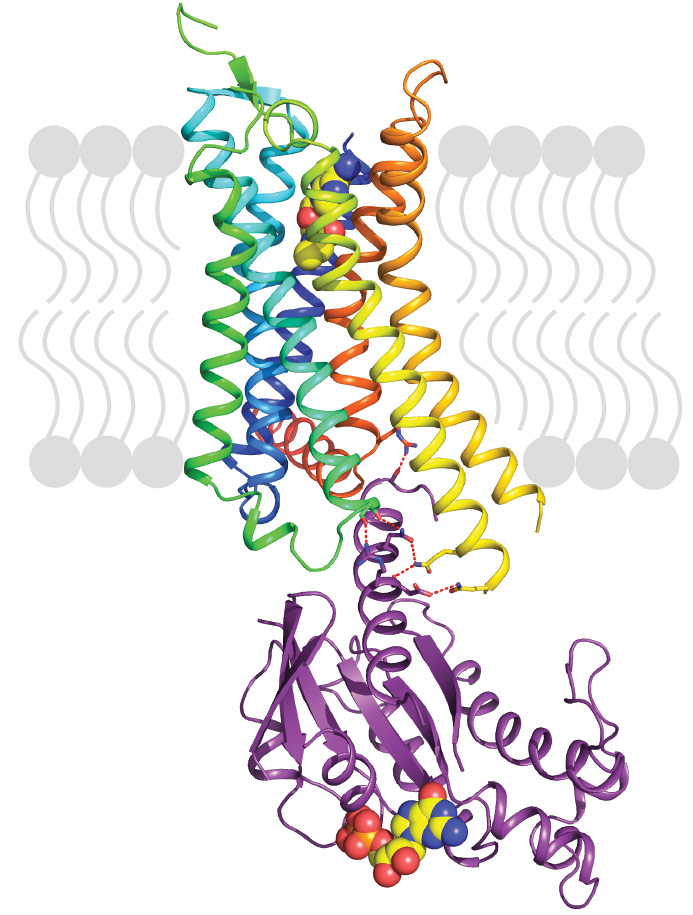 |
|
Figure 1. Structure of agonist-bound A2AR coupled to mini-Gs. The receptor is shown in rainbow colouration (N-terminus blue, C-terminus red) and is bound to the agonist NECA (shown in spheres). Mini-Gs is shown in purple with specific hydrophilic contacts to A2AR shown as red dashed lines and the molecule of bound GDP shown in spheres. |
For example, the sequence at the cytoplasmic end of transmembrane helix 7 (H7) and the start of the amphipathic helix H8 is R7.56IREFR in A2AR compared to S7.56PDFRI in β2AR; all the residues labelled in bold interact with mini-Gs, but none of the equivalent residues in β2AR make contact to Gs in the crystal structure. Thus the structure of A2AR bound to mini-Gs starts to address what are the molecular determinants of specificity in GPCRs that dictate which G protein it couples to. There are only 16 different Gα subunits expressed in humans compared to over 750 GPCRs, often with multiple different GPCRs and G proteins all being expressed in the same cell, and multiple different G proteins coupling to a single GPCR. A major challenge in the field is to understand how the complex interplay between structural determinants, kinetics and sub-cellular distribution results in the signalling characteristics of GPCRs within specific cell types.
Principal publication and authors
Structure of the adenosine A2A receptor bound to an engineered G protein, B. Carpenter, R. Nehmé, T. Warne, A.G.W. Leslie and C.G. Tate, Nature 536, 104-107 (2016); doi: 10.1038/nature18966.
MRC Laboratory of Molecular Biology, Cambridge (UK)
References
[1] Carpenter & Tate, Protein Eng. Des. Sel., 1-11 (2016) doi: 10.1093/protein/gzw049.
Acknowledgements
This work was supported by a grant from Heptares Therapeutics Ltd, the ERC (grant EMPSI 339995) and core funding from the Medical Research Council (MC_U105197215 and MC_U105184325). We thank the beamline staff at the European Synchrotron Radiation Facility (beamlines ID23-2, ID30-A3 and ID29) and at Diamond Light Source (beamline I24).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.