- Home
- News
- Spotlight on Science
- Transfer hydrogenation...
Transfer hydrogenation catalyst deactivation solved through in situ X-ray absorption spectroscopy
05-08-2015
Scientists from University of Leeds (UK) and the XMaS CRG beamline have developed a new approach to studying complex immobilised catalysts using a combination of X-ray absorption spectroscopy at both hard and soft X-ray energies and lab-based physical organic chemistry. This strategy was successfully demonstrated with an immobilised hydrogen transfer catalyst, showing that the slow deactivation of this catalyst was due to gradual exchange of a chloride ligand on the Ir catalyst with an alkoxide. This led to an observable loss of chloride content and a build-up in potassium content in the catalyst as the reaction progressed. The innovative measurements at the ESRF were crucial in identifying the catalyst deactivation pathway.
Transfer hydrogenation is a ubiquitous class of catalytic reaction in chemical syntheses [1]. Not only do these reactions eliminate the need for specialised hydrogenation equipment when scaled up, they also proceed with excellent selectivity under mild conditions. In addition, transfer hydrogenation forms part of the ‘borrowing hydrogen’ mechanism, a highly useful concept in modern synthetic methodology [2]. The main catalytic cycles in transfer hydrogenation using Ru/Ir catalysts are generally well understood. However, direct insights into deactivation of these catalysts, which are critical for maximising their efficiency, are rare. The elaborate nature of the most active catalysts also means that catalyst recovery is important in industrial application.
We recently reported an immobilised transfer hydrogenation catalyst [Cp**IrCl2]2, (Cp** = immobilised 1,2,3,4,5-pentamethylcyclopentadienyl ligand) on a polymer support, which was reused up to 30 times with little leaching of iridium (<3%, see Figure 1) [3]. This was achieved through a novel immobilisation strategy whereby the catalyst is linked to the support through a strongly bound Cp* ligand. A slow and partial deactivation of the catalyst was observed over 30 uses, which prompted a mechanistic study into its deactivation in order to further improve the catalyst for application in fine chemical and pharmaceutical syntheses. However, the effectiveness of traditional analytical techniques for studying this immobilised catalyst was limited, due to the lack of an NMR active label on the catalyst and interference from the support resin. A more selective tool, such as X-ray absorption spectroscopy (XAS), was required.
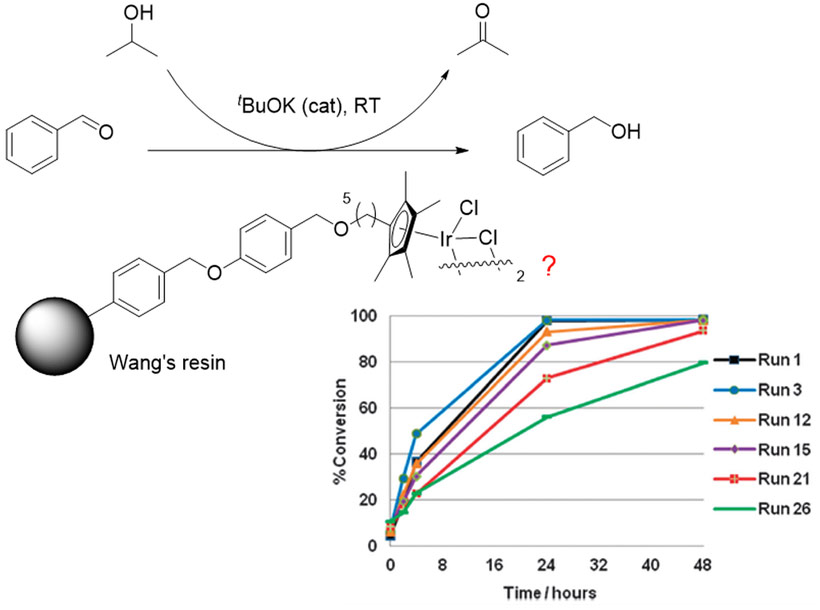 |
|
Figure 1. A novel immobilised catalyst for transfer hydrogenation which can be recycled up to 30 times. |
XAS studies on this catalyst and its deactivation were carried out at BM28 (XMaS, the UK-CRG). In contrast to the common practice of focusing on the metal centre, the team employed a holistic approach to investigate the bonding environment around Ir and Cl atoms in the ex situ catalysts (fresh, deactivated and HCl treated). This led to the crucial identification of a significant amount of potassium cations in the deactivated catalyst, which formed KCl upon treatment with aqueous HCl.
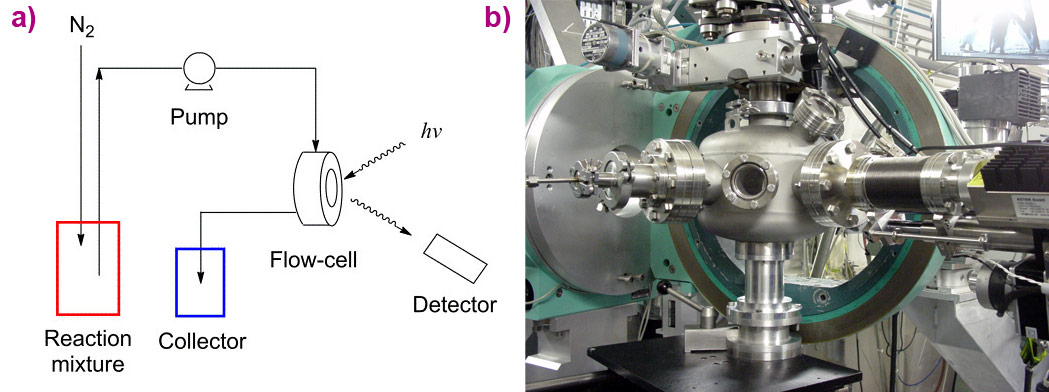 |
|
Figure 2. a) Schematic of the XAS setup for in situ fluorescence XANES at low energy; b) Flow cell mounted inside an in-vacuum sample chamber on the XMaS diffractometer. The spectra were collected with a silicon drift detector (Ketek). |
These preliminary results culminated in an in situ experiment employing a spectroscopic flow-cell (see Figure 2), in which the slow loss of catalytic activity over time was directly linked with the loss of chloride ligands from the iridium catalyst. Monitoring XANES (X-ray absorption near-edge spectroscopy) spectra of the catalyst (see Figure 3) led to the observation of a concurrent increase in potassium content over the same timescale with catalyst deactivation, which happened as the chloride ligands were replaced with alkoxides to generate a less active Cp*Ir-trialkoxide complex (see Figure 4).
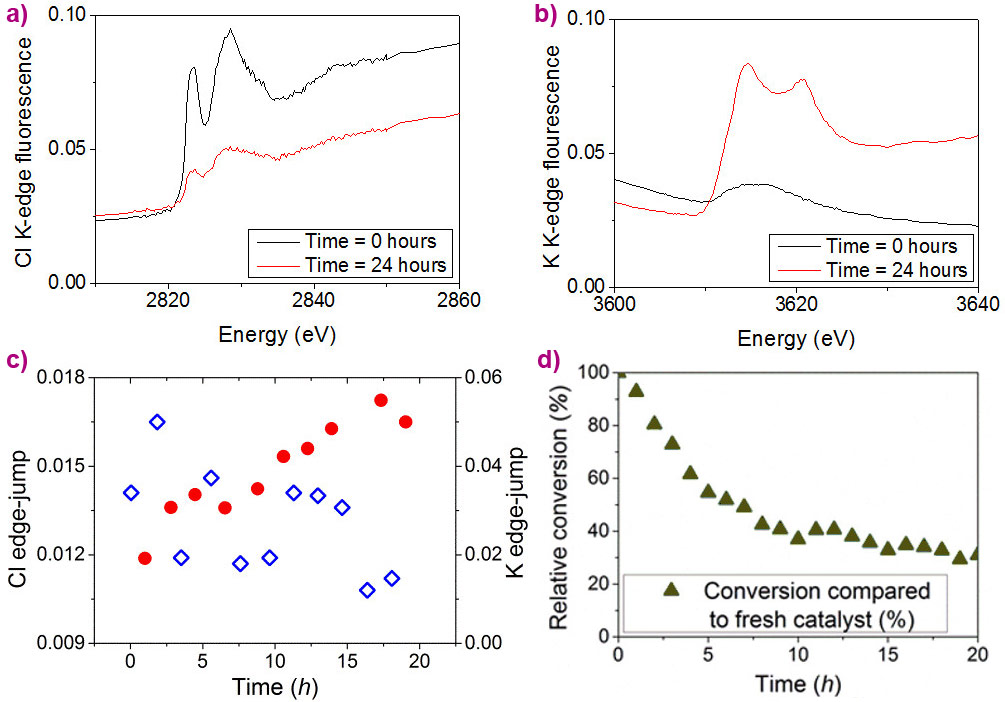 |
|
Figure 3. Fluorescence XANES spectra measured across the Cl K-edge (a) and K K-edge (b) at time = 0 and 24 hours; (c) Cl K-edge jump (blue) and K K-edge jump (red) under turnover conditions; (d) Catalytic conversion over time relative to fresh precatalyst by GC. |
Appropriate re-activation strategies have consequently been developed based on these findings to regenerate the catalyst to its original reactivity through reinstatement of chloride ligands in the catalyst. In addition, changes to the reaction conditions can potentially suppress the undesirable ligand exchange of chloride. Importantly, the study showcased the potential of this new multi-element XAS approach in studying solution based catalysis and in this particular case even with an immobilised catalyst.
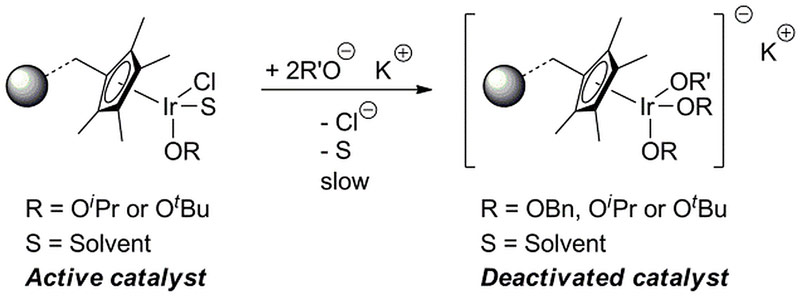 |
|
Figure 4. Proposed deactivation pathway of the immobilised catalyst. |
Principal publication and authors
Activation and deactivation of a robust immobilized Cp*Ir-transfer hydrogenation catalyst: a multielement in situ X-ray absorption spectroscopy study, G.J. Sherborne (a), M.R. Chapman (a), A.J. Blacker (a), R.A. Bourne (a), T.W. Chamberlain (b), B.D. Crossley (c), S.J. Lucas (a), P.C. McGowan (a), M.A. Newton (d), T.E.O. Screen (c), P. Thompson (d), C.E. Willans (a), B.N. Nguyen (a), J. Am. Chem. Soc. 137, 4151 (2015).
(a) Institute of Process Research & Development, School of Chemistry, University of Leeds (UK)
(b) School of Chemistry, University of Nottingham (UK)
(c) Yorkshire Process Technology, Leeds Innovation Centre (UK)
(d) XMaS (BM28), ESRF
.
References
[1] T. Ikariya, A.J. Blacker, Acc. Chem. Res. 40, 1300 (2007).
[2] T.D. Nixon, M.K. Whittlesey, J.M.J. Williams, Dalton Trans. 753 (2009).
[3] S.J. Lucas, B.D. Crossley, A.J. Pettman, A.D. Vassileiou, T.E.O. Screen, A.J. Blacker, P.C. McGowan, Chem. Commun. 49, 5562 (2013).
Top image: In situ X-ray absorption spectroscopy confirms catalyst deactivation by loss of chlorine (blue) and increase in potassium (red).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.