- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Structure of materials
- Guest species switches negative and positive thermal expansion in YFe(CN)6
Guest species switches negative and positive thermal expansion in YFe(CN)6
The presence of guest ions (K+) and molecules (H2O) can switch thermal expansion of YFe(CN)6 from negative (αv = –33.67 x 10*-6 K* -1) to positive (αv = +42.72 x 10*-6 -1), a range that covers thermal expansion of most inorganic compounds. This presence has a critical damping effect on transverse vibrations, thus inhibiting negative thermal expansion.
The control of thermal expansion in NTE materials is mainly achieved by chemical substitution. Since chemical substitution can have a pronounced effect on electronic structure and crystal structure, thermal expansion can be well controlled, especially in electronic-driven NTE materials, such as PbTiO3-based ferroelectrics [1]. However, in open-framework materials, chemical substitution might not be a direct method to adjust thermal expansion, because NTE is associated with the lattice dynamics rather than the electronic structures. For example, the linear coefficient of thermal expansion (CTE, αl) for Zr1–xMxW2O8–y (M = Sc, In, Y) materials varies only over a narrow range (-7.3 to -8.7 x 10-6 K-1) [2].
 |
|
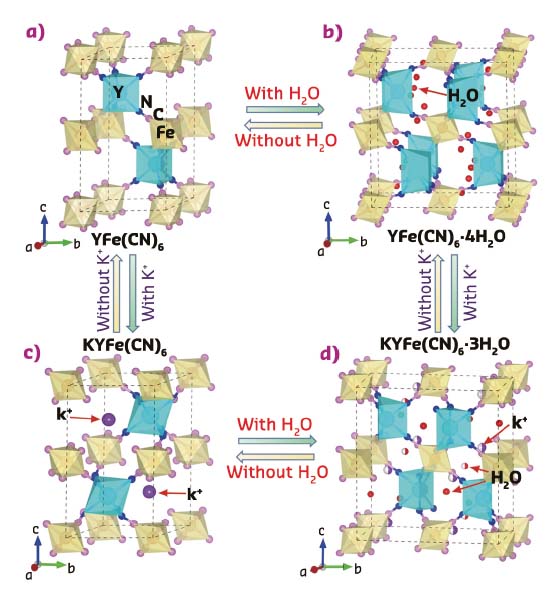
Fig. 137: Crystal structure of YFe(CN)6-based Prussian blue analogues with or without guest K+ ions and H2O molecules. a) YFe(CN)6 (P63/mmc), b) YFe(CN)6 × 4H2O (Cmcm), c) KYFe(CN)6 (P-31c) and d) KYFe(CN)6×3H2O (Pbnm). FeC6 and YN6 polyhedra are in yellow and light blue colour, respectively. |
Figure 137 shows a YFe(CN)6 entire structure with the YN6 and FeC6 groups bridged through CN units, and the guest molecules (H2O) or ions (K+) in the empty spaces of its framework structure. When YFe(CN)6 is without any guest ions or molecules, a strong NTE can be observed (αv = –33.7 x 10–6 K–1). It is intriguing that thermal expansion of YFe(CN)6 can be largely switched between negative and positive via the effect of guest K+ ions and H2O molecules (Figure 138a).
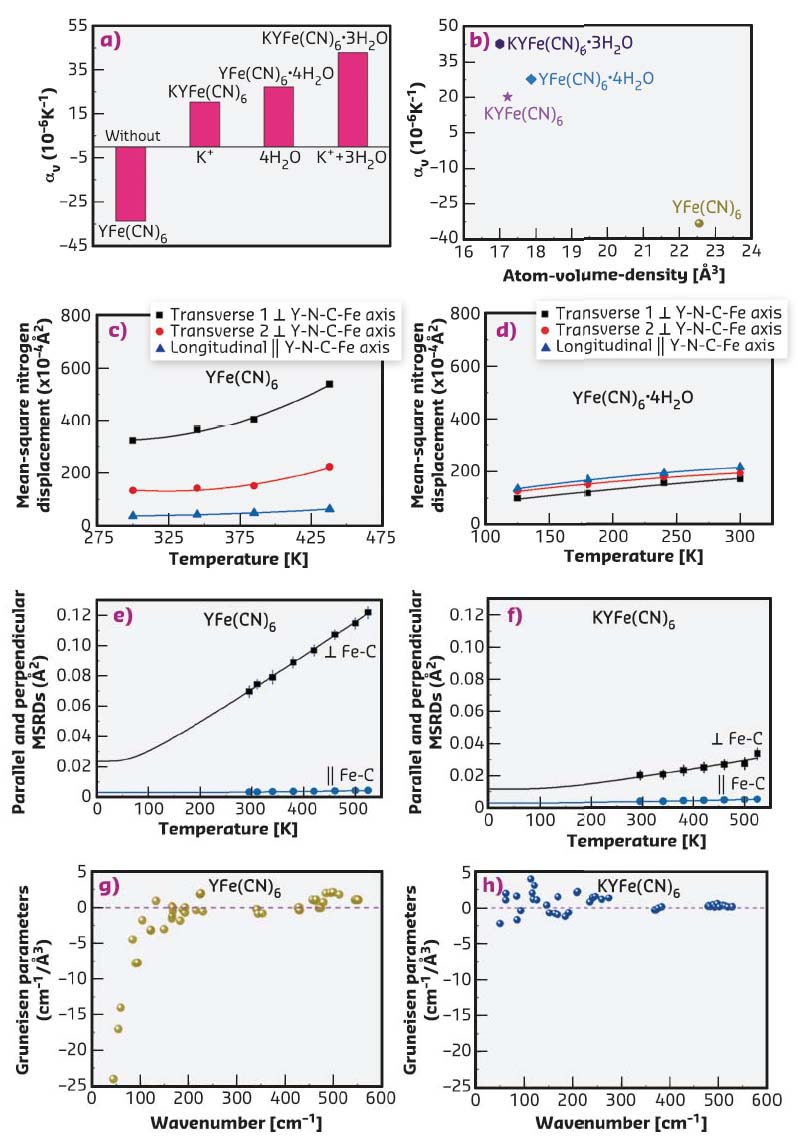
In this work, the detailed mechanism for the role of guest ions and molecules on the switched thermal expansion was investigated. Here, the phenomenon was interpreted according to both structure and lattice dynamics by joint studies using neutron powder diffraction, extended X-ray absorption fine structure (EXAFS) at beamline BM08-LISA, and first-principle calculations (Figures 138b-h). Firstly, Figure 138b shows correlation between atom-volume-density and CTE, that is, if there is enough space in the open-framework structure, the NTE arises due to possibly strong transverse vibrations of bridging atoms. Otherwise, the transverse vibrations are reduced and thus NTE should be restrained or switched to PTE. Secondly, there are strong anisotropic atomic displacement parameters (ADPs) in YFe(CN)6 (Figure 138c), in which the transverse ADPs of both N and C atoms are much larger than those of longitudinal ones.
 |
|
Fig. 138: a) CTE, αv, in YFe(CN)6 and related compounds. b) Correlation between atom-volume-density and αv. Temperature variation of ADPs of the N atoms of c) YFe(CN)6 and d) YFe(CN)6 × 4H2O, respectively. Perpendicular (squares) and parallel (circles) MSRDs of the Fe-C atomic pairs measured by EXAFS in e) NTE YFe(CN)6 and f) PTE KYFe(CN)6. The solid lines are the corresponding best-fit with the Einstein model. The mode Grüneisen parameters of g) NTE YFe(CN)6 and h) PTE KYFe(CN)6 as a function of frequency. |
However, after the insertion of guest H2O molecules, the ADPs are nearly identical for both transverse and longitudinal ones (Figure 138d), which means that the transverse Fe-C and Y-N thermal vibrations are much hindered. Thirdly, the atomic mean-square relative displacements (MSRDs) determined by Fe K-edge EXAFS measurements indicates that there is much larger transverse Fe-C vibrations in NTE YFe(CN)6 than in PTE KYFe(CN)6 (Figures 138e-f). Finally, from Figures 138g-h, it can be seen that most vibrational modes with negative Grüneisen parameters in YFe(CN)6 switch to positive when K+ ions are inserted, thus resulting in PTE of KYFe(CN)6. The study demonstrates a potential approach to achieve adjustable CTE for those NTE framework materials and other multifunctional materials.
Principal publication and authors
Switching Between Giant Positive and Negative Thermal Expansions of a YFe(CN)6-based Prussian Blue Analogue Induced by Guest Species, Q. Gao (a), J. Chen (a), Q. Sun (b), D. Chang (b), Q. Huang (c), H. Wu (c), A. Sanson (d), R. Milazzo (d), H. Zhu (a), Q. Li (a), Z. Liu (a), J. Deng (a) and X. Xing (a), Angew. Chem. Int. Ed. 56, 9023 (2017); doi: 10.1002/anie.201702955.
(a) Department of Physical Chemistry, USTB (China)
(b)School of Physics and Engineering, ZZU (China)
(c) NIST Center for Neutron Research (USA)
(d) Department of Physics and Astronomy, University of Padova (Italy)
References
[1] J. Chen et al., J. Am. Chem. Soc. 130, 1144 (2008).
[2] N. Nakajima et al., Solid State Commun. 128, 193 (2003).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.