- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Structural biology
- Structures of PAR2 complexes reveal new modes of modulation
Structures of PAR2 complexes reveal new modes of modulation
Synchrotron radiation has been used to reveal the structure of a cell surface receptor involved in pain regulation. This has revealed new opportunities for the development of agents for pain relief.
Protease-activated receptors (PARs) are implicated in several diseases including inflammation, thrombosis, pain and cancer. These receptors are members of the G protein-coupled receptor (GPCR) family that reside in the cell membrane and include the targets for many medicines. However, PARs are unusual in not being directly activated by agonists circulating in the extracellular environment. Rather, activation occurs through extracellular proteases, which remove a short section of the polypeptide chain of the receptor, and the new end of the polypeptide chain acts as the agonist by inserting into and activating the receptor (Figure 24). This means of activation is commonly referred to as ‘tethered agonism’ and it can be mimicked by the addition of peptide corresponding to the sequence of the end of the receptor. Tethered agonists present challenges for the discovery of antagonists (which block the response), as, after proteolysis, the agonist is always adjacent to the receptor activation site, and its local concentration is far higher than for a conventional non-tethered system [1]. Despite the involvement of PARs in several diseases, and being the subject of drug discovery efforts in many companies, only one medicine that targets a protease-activated receptor has received regulatory approval for use in patients. Vorapaxar, which is licensed for use in patients with a risk of specific cardiovascular events, is an antagonist of protease-activated receptor 1 [2].
 |
|
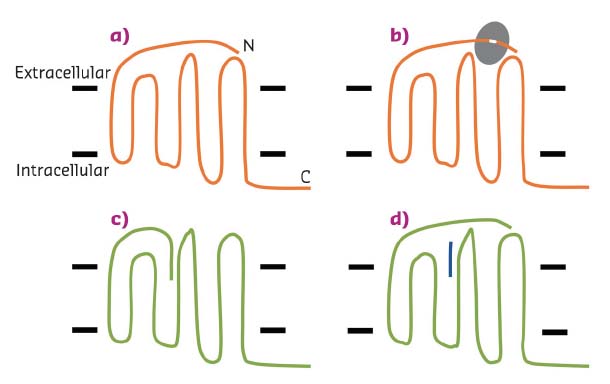
Fig. 24: Schematic representation of the activation on PARs. a) An inactive PAR (orange); the N and C-termini of the protein are labelled, and the black bars indicate the approximate location of the cell membrane. b) A protease enzyme (grey) binds to the PAR, removing a section of the N-terminus. c) The newly generated N-terminus of the PAR binds to a pocket and activates the receptor (green). d) Activation of the receptor can be mimicked without a protease by addition of a short peptide (blue). |
AstraZeneca and Heptares Therapeutics embarked on a joint project to discover antagonists of protease-activated receptor 2 (PAR2) for potential development of analgesic agents. Two series of antagonists were identified from screening a library of drug-like compounds and a library of DNA-encoded molecules. Using the Heptares stabilised receptor (StaR) technology, crystal structures for the PAR2, bound to a representative of each of these series, were determined. These clearly show the binding site for one antagonist on the outside of the receptor and a site for the other adjacent to the presumed pocket for the tethered agonist. Each molecule appears to function as an antagonist by preventing the conformational changes required for the tethered agonist to bind to and activate the receptor. Using ID29, a structure was also obtained for PAR2 bound to a small molecule antagonist and a fragment from the monoclonal antibody MAB3949 (Figure 25). Loops of the heavy and light chains of the antibody bind to the extracellular face of PAR2. The epitope is comprised of the amino terminus, extracellular loops 2 and 3, and the extracellular end of trans-membrane helix 6. This mode of antibody binding is different from examples where the antibody targets the site that is cleaved by proteases before receptor activation. This is mirrored in assay results where the latter antibodies are able to prevent activation of the receptor after protease cleavage, but are unable to block activation by the addition of peptides. MAB3949 can prevent activation in both cases, possibly through direct competition for access to the agonist binding site.
 |
|
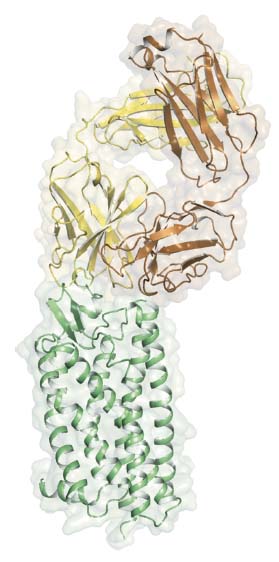
Fig. 25: The structure of PAR2 (green) in complex with FAB3949 (heavy chain in yellow and light chain in brown). The FAB binds to the extracellular side of the receptor and the observed interaction provides a structural rationale for the antagonist activity of MAB3949. |
The structural work has demonstrated three new means by which PARs can be antagonised and may pave the way for further developments for this difficult sub-group of targets.
Principal publication and authors
Structural insight into allosteric modulation of protease-activated receptor 2, R. Cheng (a), C. Fiez-Vandal (a), O. Schlenker (a), K. Edman (b), B. Aggeler (c), D.G. Brown (b), G. Brown (a), R. Cooke (a), C. Dumelin (d), A. Doré (a), S. Geschwindner (b), C. Grebner (b), N-O. Hermansson (b), A. Jazayeri (a), P. Johansson (b), L. Leong (c), R. Prihandoko (a), M. Rappas (a), H. Soutter (d), A. Snijder (b), L. Sundström (b), B. Tehan (a), P. Thornton (b), D. Troast (d), G. Wiggin (a), A. Zhukov (a), F. Marshall (a) and N. Dekker (b), Nature 545, 112 (2017); doi: 10.1038/nature22309.
(a) Heptares Therapeutics (UK)
(b) AstraZeneca (Sweden, USA and UK)
(c) Bio-techne (USA)
(d) X-Chem Inc. (USA)
References
[1] R. Ramachandran et al., Nat Rev Drug Discov. 11, 69 (2012).
[2] B. E. Hawes et al., Eur J Pharm. 762, 221 (2015).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.