- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2017
- Structural biology
- DNA methylation affects binding of most human transcription factors
DNA methylation affects binding of most human transcription factors
The first global analysis of the effect of cytosine methylation on DNA binding specificities of human transcription factors has been carried out. The data revealed that methylation of CpG dinucleotides can influence DNA-binding of most transcription factors – often positively – and identified the molecular mechanisms behind it.
The role of both transcription factors (TFs) and DNA methylation in epigenetic inheritance is well established, and many principal mechanisms controlling gene expression are well understood. However, as the methylation changes DNA structure, it has the potential to affect binding of all TFs. To characterise this effect, the binding specificities of human TFs were analysed towards unmethylated and CpG-methylated DNA [1, 2]. Binding of most major classes of TFs, including bHLH, bZIP, and ETS, was inhibited by mCpG. In contrast, TFs such as homeodomain, POU, and NFAT proteins preferred to bind to methylated DNA.
Structural analysis of homeodomain proteins, using diffraction data collected at ID29 and ID23-1, was used to identify the molecular mechanisms behind the preference of these TFs towards mCpG. The crystal structures of HOXB13 DBD, bound to dually methylated versions of its preferred site CTCGTAAA in the presence or absence of its heterodimeric partner MEIS1, were solved. Two regions of HOXB13 interact with DNA: the recognition helix α3, which tightly packs into the major groove, and the N-terminal tail interacting with the minor groove (Figure 13a). Analysis of the TF-DNA contacts showed that HOXB13 recognises mCpG by direct hydrophobic interactions between amino acids and the 5-methyl groups of both methylcytosines of the CpG dinucleotide: Ile262 forms a hydrophobic contact with the first methylcytosine, whereas Val269 recognises the second methylcytosine opposite to the guanine of the TCG sequence (Figure 13a). In addition, the aliphatic chain of Arg258 interacts with Ile262 and contributes to the hydrophobic environment of this region. These hydrophobic interactions were also present in the HOXB13:MEIS1-DNA crystal structure, indicating that the methyl groups of both cytosines are robustly recognised by HOXB13 in multiple physiologically relevant contexts (Figure 13b).
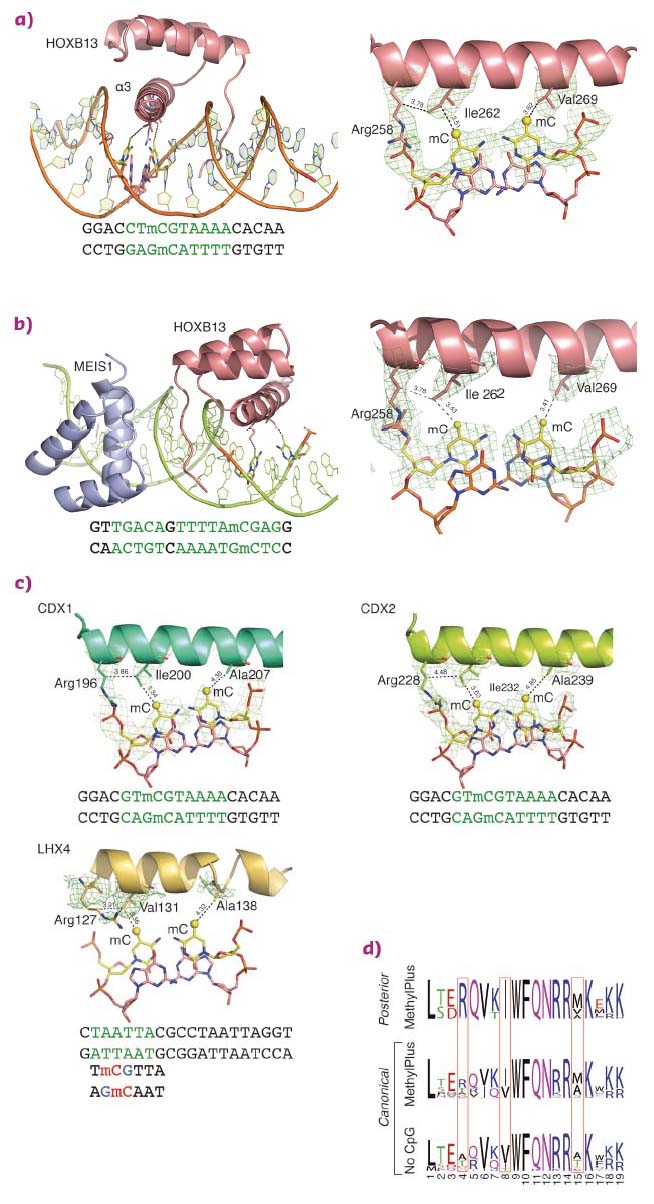 |
|
Fig. 13: Molecular basis of recognition of mCpG by homeodomain proteins. |
To determine whether the mechanism of recognition of mCpG by homeodomains is general, the crystal structures of three additional proteins: CDX1, CDX2, and LHX4 were solved in their DNA-bound forms (Figure 13c). The crystal structures of CDX1 and CDX2 bound to their preferred GTmCGTAAA subsequence indicated that they directly recognise the 5-methyl group of methylcytosine by using amino acids in the same relative positions (Figure 13c). While the paraHOX proteins CDX1 and CDX2 bind strongly to mCpG in their TmCGTAAA motif, LHX4 binds to the canonical TAATTA motif and displays somewhat weaker binding to the mCpG-containing sequence TmCGTTA. The structure of LHX4 bound to a canonical TAATTA motif showed hydrophobic residues Val131 and Ala138 in positions suitable for the formation of hydrophobic contacts with methylcytosines. The aliphatic chain of Arg127 also supports a hydrophobic interaction (Figure 13c). These three residues are conserved in all LHX proteins, explaining their general preference towards mC. In contrast, in DLX3, a homeodomain that shows a much weaker preference for mC, the key residues corresponding to Arg127 and Ala138 were replaced by a threonine and a serine, respectively, leading to a decrease in hydrophobicity (Figure 14a). Furthermore, in TLX2, which does not bind to TmCGTTA, hydrophobicity of the entire binding site was lost (Figure 14b) [3].
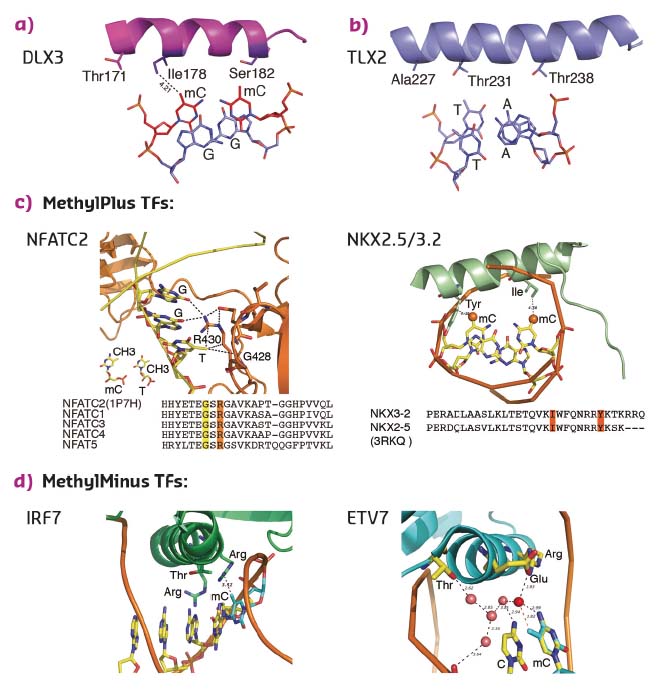 |
|
Fig. 14: Structural basis of TFs preferring methylated (a, c) or unmethylated (b, d) CpG. |
Analysis of the amino acid sequences of different homeodomains that either do or do not bind to mCpG-containing sites confirmed the critical role in mCpG recognition of the three residues located at the beginning and end of the recognition helix (Figure 13d). Analysis of structures of the methyl-plus TFs, including some NKX- [4, 5] and NFAT-family proteins, confirmed that the preference for methylcytosine is based on hydrophobic interactions with the 5-methyl group (Figure 14c). Structural analyses also confirmed the mechanism by which methylation of cytosine inhibits TF-DNA binding; in all cases examined, the negative impact of methylation was due to steric hindrance (Figure 14d).
The discovery that many developmentally important TFs prefer to bind to methylated CpG sites, together with the mechanistic understanding of the increased affinity, forms a solid basis for future analyses of the role of DNA methylation on cell differentiation, chromatin reprogramming and transcriptional regulation.
Principal publication and authors
Impact of cytosine methylation on DNA binding specificities of human transcription factors, Y. Yin (a), E.Morgunova (a), A. Jolma (a), E. Kaasinen (a), B. Sahu (b), S. Khund-Sayeed (c), P.K. Das (b), T. Kivioja (b), K. Dave (a), F. Zhong (a), K.R. Nitta (a), M. Taipale (a), A. Popov (d), P.A. Ginno (e), S. Domcke (e,f), J. Yan (a), D. Schübeler (e,f), C. Vinson (c) and J. Taipale (a,b), Science 356, 6337, eaaj2239 (2017); doi:10.1126/science.aaj2239.
(a) Division of Functional Genomics and Systems Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm (Sweden)
(b) Genome-Scale Biology Program, University of Helsinki (Finland)
(c) Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland (USA)
(d) ESRF
(e) Friedrich-Miescher-Institute for Biomedical Research (FMI), Basel (Switzerland)
(f) Faculty of Science, University of Basel (Switzerland)
References
[1] A. Jolma et al., Genome Res. 20, 861-873 (2010).
[2] A. Jolma et al., Nature 527, 384-388 (2015).
[3] K. Miyazono et al., Embo J. 29, 1613-1623 (2010).
[4] L. Pradhan et al., Biochemistry 51, 6312-6319 (2012).
[5] M. J. Giffin et al., Nat. Struct. Biol. 10, 800-806 (2003).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.