- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2015
- Structure of materials
- Structure and properties of complex hydride perovskite materials
Structure and properties of complex hydride perovskite materials
Borohydride perovskites pave the way to functional design in complex hydride materials for energy-applications. A novel mechanism is suggested for symmetry-design in perovskites, arising from the hydridic nature of hydrogen and inverting the temperature-dependency of lattice instabilities.
Perovskite materials are ubiquitous in materials science because of their distinct adjustability of physical properties and the readiness to incorporate most chemical elements from the periodic table. Slight alterations of the crystal structure can induce dramatic changes in the physical properties. Widely known as reducing agents in organic chemistry, the high hydrogen content per unit mass/volume and the rich structural dynamics of metal borohydrides have triggered vast investigations on their energy-related implementation as materials for solid-state hydrogen storage and all-solid batteries. Initially this was driven by the potential to develop LiBH4 into a storage medium for on-board automobile applications. The motivation behind metal borohydrides in the materials science community hence directly targets a prime issue of modern consumer societies.
Due to the hydridic nature of hydrogen in the BH4-molecule, borohydrides are somewhat an outsider, chemically speaking. The covalently bound hydrogen in molecular chemistry commonly carries a positive formal charge (protic hydrogen).
Despite the application-oriented search for novel complex hydrides, the rational structure-property design of metal borohydrides is still in its infancy. The principal objective of this study was to provide an extensive characterisation of a host material capable of meeting the requirements of genuine functional design, in order to take borohydrides beyond hydrogen storage applications in the future. Ideally, such a host material is of reduced complexity and superior stability.
Motivated by our previous discovery of a double-cation borohydride KMn(BH4)3 crystallising in the perovskite lattice, this structure type was systematically explored leading to the discovery of over 45 different materials whose formability criteria were evaluated based on the commonly used Goldschmidt tolerance factor (Figure 44).
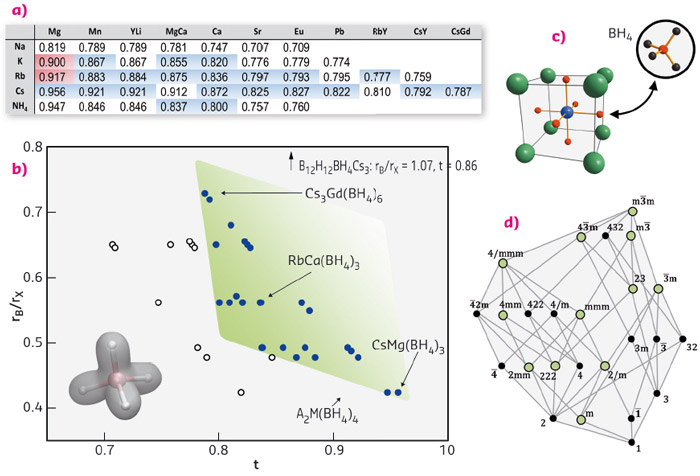 |
|
Fig. 44: (a-b) Formability field (green) of borohydride perovskites based on octahedral vs. tolerance factor t (listed in table (a). The AM(BH4)3 structure is shown in (c). (d) Sub-groups of the cubic aristotype, green circles correspond to crystal symmetries of discovered borohydrides. |
We began to study this field on powder samples prepared by mechanochemistry using the Dectris Pilatus M2 detector at BM01A, systematically heating the sample to its decomposition (detection of thermal stability). With the resulting materials, we have managed to produce metal borohydrides with unforeseen physical properties such as photoluminescence, magnetic refrigeration, semiconductivity, and proton-hydride interactions between cations NH4+ and anions BH4– which facilitate H2-formation. Simple concepts such as the tolerance factor and ionic substitution were applied in order to obtain a desired property, following the established approaches for metal-oxide perovskites.
An unexpected structural trend was revealed for metal-borohydride perovskites. For perovskites in general, both the rotation (tilt) of MO6 octahedra and atomic displacements tend to vanish upon heating due to an increase in crystal symmetry, likewise for polar physical properties, such as an electric dipole moment. But for AM(BH4)3 (A: alkali metal, M: bivalent metal), we often find this trend to be reversed, hence stabilising low crystal symmetry at high temperature, and providing the structural requirements for polar properties. Our working hypothesis assigns the origin of this behaviour to interactions between molecular B-H vibrations and lattice vibrations and hence proposes a novel mechanism useful for tailoring perovskite symmetries [1]. For instance, a prominent lattice instability (R-point) of symmetry Pnma is activated upon heating KCa(BH4)3 into the HT-phase (Figure 45). A discrete step in the bandwidth of the Raman B-H stretching signature concurs with the structural transition, suggesting that internal modes of the BH4 molecule and lattice modes stabilising the crystal symmetry communicate with each other.
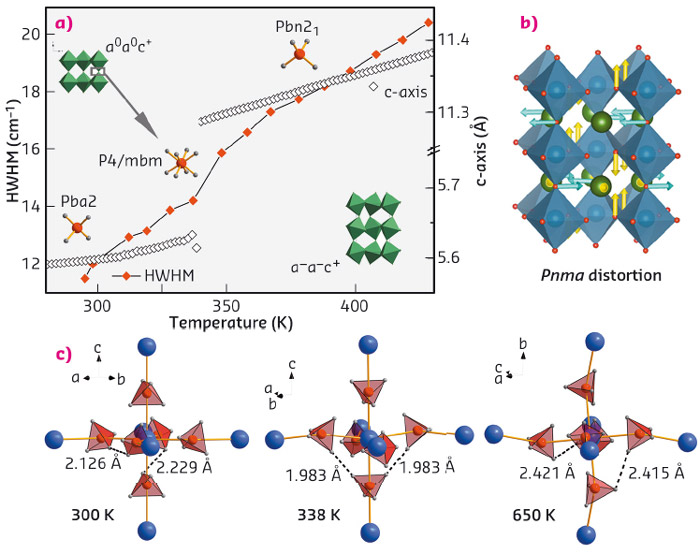 |
|
Fig. 45: (a) Changes in structure during the high-temperature phase transition of KCa(BH4)3, followed by in situ powder diffraction and Raman spectroscopy; (b) The corresponding apolar parent distortion; (c) relevant short H···H contacts within the Ca(BH4)6-octahedron. |
Theoretical solid state calculations provided further insight into the transformation mechanism, pointing towards the crucial role of close homopolar repulsive di-hydrogen for the structural behaviour of AM(BH4)3 (Figure 45). As well as providing a stable host for functional design, the borohydride perovskite hence incorporates weak interactions, which define structure and packing in molecular and supramolecular chemistry, into a structural behaviour otherwise controlled by lattice vibrations. Meanwhile, the role of these interactions in AM(BH4)3 has been verified by quasielastic neutron scattering studies of BH4 reorientations [2] and single crystal X-ray diffraction performed at BM01A on the first double-cation metal-borohydride single crystal [3].
Principal publication and authors
P. Schouwink (a), M.B. Ley (b), A. Tissot (c), H. Hagemann (c), T.R. Jensen (b), L. Smrčok (d) and R. Černý (a), Nature Comm. 5, 5706 (2014); doi: 10.1038/ncomms6706.
(a) Laboratory of Crystallography, Department of Quantum Matter Physics, University of Geneva (Switzerland)
(b) Interdisciplinary Nanoscience Center (iNANO), Department of Chemistry, University of Aarhus (Denmark)
(c) Department of Physical Chemistry, University of Geneva (Switzerland)
(d) Institute of Inorganic Chemistry, Slovak Academy of Sciences, Bratislava, (Slovak Republic)
References
[1] J.M. Rondinelli and C.J. Fennie, in Adv. Mater. 24, 1961-1968 (2012).
[2] P. Schouwink et al., in J. Phys. Condens. Matt., 27, 265403 (2015).
[3] P. Schouwink et al., in CrystEngComm, 17, 2682-2689 (2015).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.