- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2015
- Structural biology
- Deciphering enzyme evolution in antibiotic biosynthesis pathways
Deciphering enzyme evolution in antibiotic biosynthesis pathways
Soil bacteria Streptomyces are the source of many biologically active compounds. The molecular basis of the evolution of a classical methyltransferase into an unusual hydroxylase in the biosynthetic pathways of two anthracycline anticancer agents is the focus of this study.
Streptomyces soil bacteria are competent chemists that are able to produce thousands of chemically complex natural products. Key to the development of this rich source of metabolites appears to be an evolutionary pressure that promotes chemical diversity; new biosynthetic pathways are continuously being formed in these bacteria, which may result in the appearance of a novel bioactive compound that provides significant competitive advantage to the producing organism. This is reflected in the biosynthetic enzymes that have been able to evolve relatively freely and, as a consequence, have resulted in similar proteins having functions that may greatly differ. In recent years, our work has focused on understanding the details of this process [1, 2], in order to facilitate future protein engineering efforts for the generation of improved bioactive natural products.
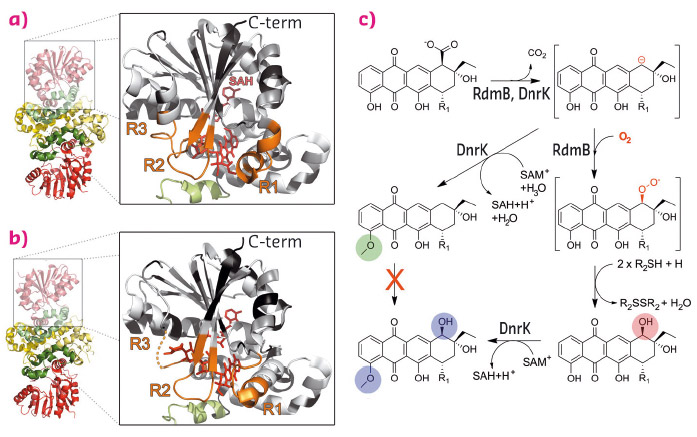
Here we have elucidated the molecular basis of the evolution of a classical methyltransferase into an unusual hydroxylase in the biosynthetic pathways of two anthracycline anticancer agents. DnrK from the daunorubicin pathway [3] is a canonical 4-O-methyltransferase, whereas the closely related RdmB (52% sequence identity) from the rhodomycin pathway is an atypical 10-hydroxylase [4] that requires SAM, a thiol reducing agent, and molecular oxygen for activity (Figure 129).
 |
|
Fig. 129: Similar structures, distinct functions. Structures of (a) DnrK and (b) RdmB with regions investigated in this study highlighted in orange (R1, R2, R3). (c) Mechanistic proposal for the divergent reactions of DnrK and RdmB. |
In order to identify the regions responsible for the functional differentiation, we selected three regions (Figure 129, R1-R3) based on the crystal structures of the enzymes. We then created chimeric enzymes by interchanging these 10 to 12 amino acid segments both individually and in different combinations. Activity assays revealed that incorporation of the R1 region from RdmB into the DnrK scaffold was critical for introduction of the 10-hydroxylation activity. Further sequence analysis revealed that RdmB contained an insertion of one additional serine residue (S297) in this region in comparison to DnrK and, consequently, addition of this single residue was sufficient for the gain of monooxygenation activity in the DnrK-Ser mutant.
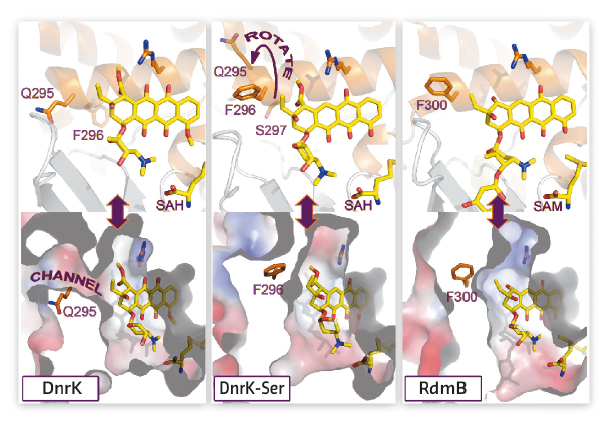
The crystal structure of DnrK-Ser in complex with aclacinomycin T and S-adenosyl-L-homocysteine was determined at beamline ID23-1 and refined to 1.9 Å resolution. The data revealed that the inserted serine S297 resides in an α-helical segment adjacent to the substrate, but in a manner where the side chain points away from the active site (Figure 130). However, at the same time the preceding phenylalanine (F296) has rotated inward adjacent to the ligand near the site of monooxygenation. Comparison of the structures of DnrK-Ser and RdmB reveals that both enzymes have a phenylalanine, F296 and F300, respectively, next to the anthracycline ligand, in contrast to native DnrK, which contains glutamine Q295 at the equivalent position. The implication of these changes is that the glutamine in DnrK allows bulk solvent to access the active site cavity via an open channel, whereas the bulkier hydrophobic phenylalanine residues block this route to the surface of the protein in DnrK-Ser and RdmB (Figure 130).
 |
|
Fig. 130: Comparison of the active site architectures of DnrK, DnrK-Ser and RdmB. |
In addition to 4-O-methylation activity, DnrK was shown to harbour a previously undetected moonlighting 10-decarboxylation activity, which we believe has been the key to the development of the 10-hydroxylation activity of RdmB. Our mechanistic proposal (Figure 129) suggests that 10-decarboxylation is a common initial step for both enzymes. In DnrK, the more open active site allows facile protonation of the carbanion by solvent molecules and results in the formation of a neutral 10-decarboxylated and 4-O-methylated anthracycline, which is unable to react further with oxygen. RdmB, on the other hand, avoids the protonation step by closing the channel to the surface of the protein and excluding solvent ions from the active site. The carbanion may be stabilised through distribution of the negative charge throughout the polyphenolic ring system and the adjacent positive charge of SAM. The delocalisation of electrons enables substrate-assisted activation of molecular oxygen and reaction of the substrate with dioxygen. Finally, the 10-peroxy anthracycline formed is reduced by a thiol reagent to generate the 10-hydroxylated end product.
Our work demonstrates that, in the evolution of antibiotic biosynthesis, addition of a single amino acid may result in the appearance of a new enzymatic reaction that leads to new bioactive products. Once this change occurred, a whole new subclass of 10-hydroxy anthracyclines became accessible to strains possessing enzymes such as RdmB in their biosynthetic repertoire.
Principal publication and authors
Divergent evolution of an atypical S-adenosyl-l-methionine-dependent monooxygenase involved in anthracycline biosynthesis, T. Grocholski, P. Dinis, L. Niiranen, J. Niemi and M. Metsä-Ketelä, PNAS 112, 9866–9871 (2015). doi: 10.1073/pnas.1501765112.
Department of Biochemistry, University of Turku (Finland)
References
[1] P. Kallio et al., Biochemistry 52, 4507–4516 (2013).
[2] P. Patrikainen et al., Chem Biol 21, 1381–1391 (2014).
[3] A. Jansson et al., J Mol Biol 334, 269–280 (2003).
[4] A. Jansson et al., J Biol Chem 280, 3636–3644 (2005).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.