- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2015
- Industrial research
- Visualising inhibition of the Nav1.7 pain channel
Visualising inhibition of the Nav1.7 pain channel
Voltage-gated sodium (Nav) channels are therapeutic targets for many diseases but isoform-selective blockers are not clinically available. We devised a new crystallisation strategy to determine structures of a previously unknown drug-binding site in the Nav1.7 channel. These results may accelerate the development of new treatments for pain and other diseases.
The opening of voltage-gated sodium (Nav) channels underlies the generation of action potentials in neurons and muscle cells. Nine Nav channel isoforms are differentially expressed throughout the human body and mutations are associated with migraine (Nav1.1), epilepsy (Nav1.1-Nav1.3, Nav1.6), pain (Nav1.7-Nav1.9), cardiac (Nav1.5), and muscle paralysis (Nav1.4) syndromes. The Nav1.7 channel is expressed primarily in sensory neurons, where gain-of-function mutations are associated with extreme pain disorders, and loss-of-function mutations cause an inability to feel pain. These contrasting phenotypes have identified Nav1.7 as a promising target in the pain perception pathway and have motivated efforts to develop selective inhibitors.
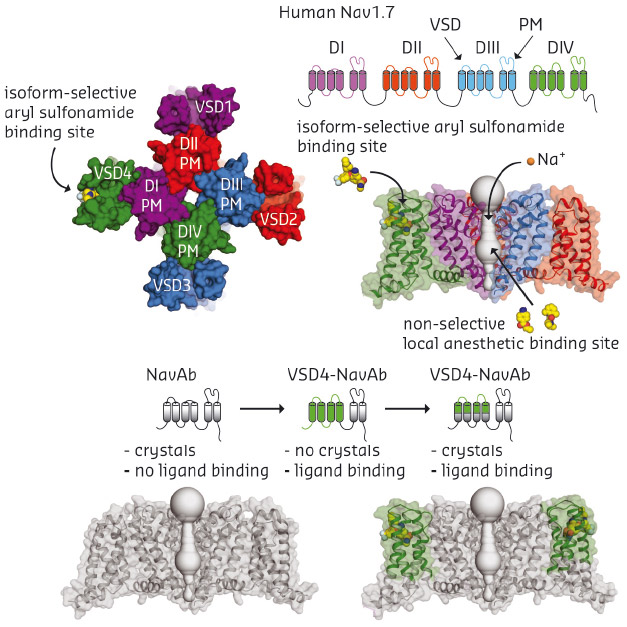
Human Nav channels are large membrane proteins (~2,000 residues) that contain a central pore module surrounded by four peripheral voltage-sensing domains (VSDs). Nav channel blockers used for the treatment of neurological and cardiovascular disorders lack isoform selectivity because they bind within the highly conserved central pore module, which limits their therapeutic utility. A recent breakthrough study identified a new class of antagonist that presumably targets a unique binding site on the fourth voltage-sensor domain (VSD4), raising hopes that an isoform-selective inhibitor can be found. Unfortunately, the inherent complexity of human Nav channels limits our ability to pursue high-resolution crystallographic studies because these proteins undergo extensive post-translational modification and associate with auxiliary subunits. To overcome the technical complexities of working with full-length human channels, we exploited a simpler homotetrameric bacterial sodium channel, called NavAb, by fusing portions of Nav1.7 VSD4 onto it. The antagonist binding-site was preserved within this VSD4-NavAb chimeric channel, and we subsequently found that this chimeric construct allowed purification, crystallisation, and structure determination of potent aryl sulfonamide inhibitors in complex with the human Nav1.7 VSD4 receptor site (Figure 139).
 |
|
Fig. 139: Overview of Nav1.7 VSD4 crystallisation strategy highlighting domain structures, 3D models, and drug binding sites in Nav channels. |
Functional studies using patch-clamp electrophysiology revealed that the inhibitors bind to an isoform-selective and extracellularly accessible site on VSD4. These inhibitors show a high level of state-dependence, potently blocking human Nav1.7 only when VSD4 is in its activated conformation. Remarkably, these inhibitors were 100,000-fold more selective for Nav1.7 over the cardiac isoform Nav1.5, a level of selectivity previously unseen among Nav channel blockers.
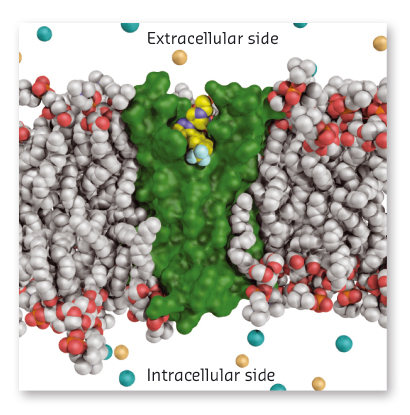
To understand the mechanism of Nav1.7 inhibition by this novel class of antagonist, we pursued high-resolution structural studies. After painstaking efforts to grow crystals, we contacted the ESRF for provision of synchrotron radiation facilities. With the aid of a dedicated staff, high-quality diffraction data were collected at beamline ID29. With access to these data, our crystallographic studies revealed that the anionic warhead from the inhibitors directly engages the fourth arginine “gating charge” on the S4 helix, which effectively traps VSD4 in its activated state. We subsequently demonstrated that isoform selectivity is achieved by inhibitor interactions with non-conserved residues found on the S2 and S3 transmembrane helices of VSD4. Remarkably, the drug receptor site is partially submerged within the membrane (Figure 140) and a peripherally bound phospholipid was observed to form a tripartite complex with the antagonist and channel.
 |
|
Fig. 140: Crystal structure of the Nav1.7 VSD4-antagonist complex viewed in a model membrane. |
In summary, we utilised a new crystallisation strategy and the ESRF to enable the structure determination of VSD4 from human Nav1.7 in complex with potent, state-dependent, isoform-selective small molecule antagonists. Mechanistically, inhibitor binding immobilises VSD4, stabilising a non-conductive state of the channel. An exposed inhibitor binding-site expands the importance of the membrane bilayer in ion channel biology. We hope that these structures will accelerate the development of new treatments for pain that target Nav1.7 and aid drug design efforts aimed at other voltage-gated ion channels.
Principal publication and authors
Structural basis of Nav1.7 inhibition by an isoform-selective small molecule antagonist, S. Ahuja (a), S. Mukund (a), L. Deng (b), K. Khakh (c), E. Chang (c), H. Ho (a), S. Shriver (a), C. Young (c), S. Lin (c), J.P. Johnson Jr. (c), P Wu (a), J. Li (d), M. Coons (a), C. Tam (a), B. Brillantes (a), H. Sampang (a), K. Mortara (a), K.K. Bowman (a), K.R. Clark (e), A. Estevez (a), Z. Xie (c), H. Verschoof (c), M. Grimwood (f), C. Dehnhardt (f), J. Andrez (f), T. Focken (f), D.P. Sutherlin (d), B.S. Safina (d), M.A. Starovasnik (a), D.F. Ortwine (d), Y. Franke (a), C.J. Cohen (c), D.H. Hackos (b), C.M. Koth (a) and J. Payandeh (a), Science 350, 6267 (2015); doi: 10.1126/science.aac5464.
(a) Department of Structural Biology, Genentech Inc., South San Francisco (USA)
(b) Department of Neuroscience, Genentech Inc., South San Francisco (USA)
(c) Department of Biology, Xenon Pharmaceuticals Inc., Burnaby (Canada)
(d) Department of Discovery Chemistry, Genentech Inc., South San Francisco (USA)
(e) Department of Biochemical and Cellular Pharmacology, Genentech Inc., South San Francisco (USA)
(f) Department of Chemistry, Xenon Pharmaceuticals Inc., Burnaby (Canada)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.