- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2013
- Structural biology
- Structure of gene regulator that plays key role in cancer
Structure of gene regulator that plays key role in cancer
Gene regulation is orchestrated by a large set of transcription factors that bind in a combinatorial fashion to DNA to regulate specific sets of genes. The ability to switch many different genes on or off using combinations of transcription factors is not only useful in the day-to-day regulation of cell function, it is also necessary for processes such as cell differentiation. Dysregulation of these processes frequently underlies diseases such as inflammation and cancer.
Although transcription factors are central to determining which genes become active, they alone are not sufficient to do the job. Cellular DNA in our cells is stored inside densely packed structures called ‘nucleosomes’ which restrict the DNA’s activity. For transcription factors to activate a gene, they work in concert with enzymes called histone acetyltransferases (HATS) that transfer acetate groups to histone proteins in nucleosomes and thereby destabilise their inhibitory structure.
One histone acetyltransferase that is essential to this process is p300. As a central ‘hub’ protein, p300 allows combinatorial regulation by binding to hundreds of transcription factors allowing integration of a very large number of cellular signalling pathways, including G-protein signalling and most immune and cancer signalling pathways. Upon binding to sets of transcription factors p300 becomes active and acetylates histones to render the chromatin fibre permissive for gene transcription.
The integrity of p300 is key for cellular physiology and aberrant p300 activity contributes to pancreatic, colon, lung as well as gastric and thyroid cancer. For example, ~40% of B-cell lymphomas carry mutations in p300, many of which inactivate the enzyme resulting in cellular transformation. Many of these disease-causing mutations are clustered in the region of the protein that is responsible for histone acetylation and in domains that are responsible for nucleosome binding indicating that dysregulation of chromatin recognition and modification are central aspects of these diseases. In addition to acting as an oncoprotein by promoting tumours, p300 can also act as a tumour suppressor.
 |
|
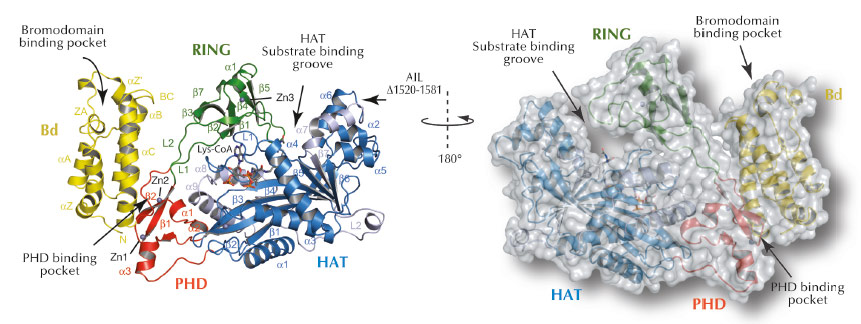
Fig. 64: Ribbon and surface representations of the crystal structure of p300 core with labelling of secondary structure elements. The Bromodomain (Bd), RING- and PHD- domains are shown in yellow, green and red, respectively. The N- and C- subdomains of the HAT domain are shown in blue and grey, respectively. Lys-CoA is shown in stick representation. |
To understand how p300 recognises and modifies chromatin and how disease mutations contribute to pathogenesis, we have determined the X-ray crystal structure of the region involved in these activities (Figure 64). Crystallising p300 was complicated by the protein’s tendency to autoacetylate itself rendering preparations of p300 structurally heterogeneous. We used enzymatic deacetylation to remove the autoacetylation and thus allow isolation of a homogeneous form. We crystallised p300 in complex with a bisubstrate inhibitor called Lys-CoA that was developed by Dr. P. Cole at Johns Hopkins University, USA.
The crystal structure of P300 revealed that the chromatin recognition domains comprising the Bromodomain (Bd), the PHD domain and the chromatin modification HAT domain adopt a compact configuration (Figure 64). We also discovered a previously unsuspected RING domain that is positioned like a ‘lid’ over the active site of the acetyltransferase. RING domains are usually E3 ligases, enzymes that catalyse transfer of ubiquitin, a small 8.5 kDa protein, to other protein substrates. Although such an activity had already been described earlier for p300, the location of the active E3 ligase domain was unknown. We found that mutations which inactivate p300 and that have been associated with its role as tumour suppressor are located in the HAT domain that acetylates chromatin. Several of the cancer causing mutations interfere with Acetyl-CoA binding or hydrolysis and inactivate the enzyme.
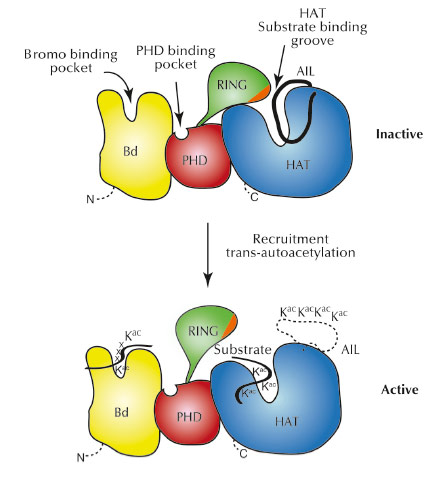
As the RING domain partially occludes the active site of the HAT domain, we believe that the RING domain has an autoinhibitory function for the enzyme (Figure 65). In agreement with this model, mutations that destabilise attachment of the RING domain result in increased p300 activity. Interestingly, a number of such destabilising RING mutations are found in different cancers indicating that p300 hyperactivity is an important aspect of these diseases. Overall, our results suggest that p300 can act not only as a tumour suppressor, but also as an oncogene, an activity that already had been suspected for this protein.
 |
|
Fig. 65: Model for p300 autoregulation and substrate acetylation. In the inactive state (top), the RING domain blocks the HAT active site. Although not observed in the present structure, we indicate, based on previous work, the docking of the autoinhibitory loop (AIL) loop to the substrate binding site of the HAT domain. |
Because of the unique chemical structure and mechanism of regulation of p300, which differs significantly from other cellular HATs, it might be possible to develop inhibitors that work specifically against this family of enzyme and that will allow development of new anti-cancer therapies.
Principal publication and authors
M. Delvecchio (a,b,c), J. Gaucher (a,b), C. Aguilar-Gurrieri (a,b), E. Ortega (a,b) and D. Panne (a,b), Nat. Struct. Mol. Biol. 20, 1040–1046 (2013).
(a) European Molecular Biology Laboratory, Grenoble (France)
(b) Unit for Virus Host-Cell Interactions, Univ. Grenoble Alpes-EMBL-CNRS, Grenoble (France)
(c) Present address: Diasorin Società per azioni, Saluggia (Italy)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.