- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2016
- Matter at extremes
- Stability of xenon oxides Xe2O5 and Xe3O2 under pressure
Stability of xenon oxides Xe2O5 and Xe3O2 under pressure
Although the most inert atomic group of the periodic table, noble gases can become reactive under extreme conditions. Diamond-anvil-cell experiments and ab initio modelling have been used to investigate a possible direct reaction between xenon and oxygen at high pressure. Two oxides with unusual stoichiometries, Xe2O5 and Xe3O2, have been synthesised at pressures above 78 GPa, pressures that exist in the Earth's interior.
In geochemistry, models of the evolution of the deep Earth and atmosphere have relied on the chemical inertness of the noble gases. These models were built using measurements of the noble gas content in the Earth’s atmosphere. However, the xenon content of the Earth’s atmosphere is anomalously low in comparison with that of stony meteorites. One theory to explain this “missing xenon paradox” is that xenon could be stored in the deep Earth. A better understanding of the chemistry of xenon under pressures relevant to the Earth’s interior (up to 3.6 million atmospheres at the centre) is needed to investigate this theory. As the binding of the valence electrons to the ionic core of a noble gas atom decreases with increasing atomic number, the heaviest noble gases (Kr, Xe) could be encouraged to form bonds using extreme conditions.
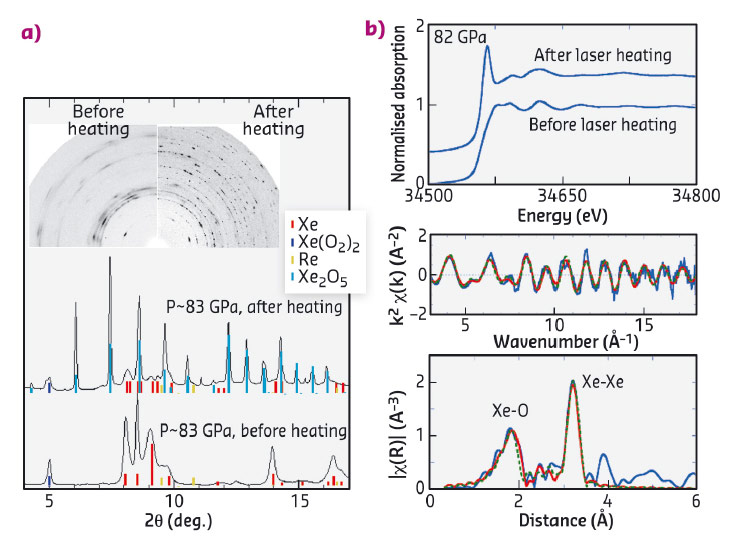
We therefore looked for possible chemical reactions under high pressure between xenon and oxygen, the most abundant element in the Earth’s mantle. Mixtures of xenon and oxygen gases were loaded into diamond anvil cells, compressed to the million-atmosphere range and heated with an infrared laser to induce reactions. We observed a reaction above 78 GPa (0.78 million atmospheres), and the products have been characterised with microfocused X-ray diffraction and X-ray absorption at the xenon K-edge at beamlines ID27 and BM23. Figure 135 shows data collected on one of these products: X-ray absorption spectroscopy demonstrated the existence of an Xe-O bond, while X-ray diffraction allowed its crystal space group and lattice parameters to be determined. Experimental data were interpreted with the help of ab initio material modelling, which also predicted new compounds that would be stable under high pressure.
 |
|
Fig. 135: X-ray diffraction and absorption patterns of ~35% Xe–65% O2 mixtures at around 82 GPa, before and after laser-heating. a) Monochromatic X-ray diffraction: the insets show the two-dimensional patterns before circular integration. Prior to laser-heating, the phases were Xe and partially amorphised Xe(O2)2. After laser-heating, the pattern consists of mostly Xe2O5 (light blue ticks). b) X-ray absorption at the Xe K-edge. The EXAFS function χ(k) and its Fourier transform χ(R) have been calculated, which enabled the determination of Xe-O and Xe-Xe distances in Xe2O5. |
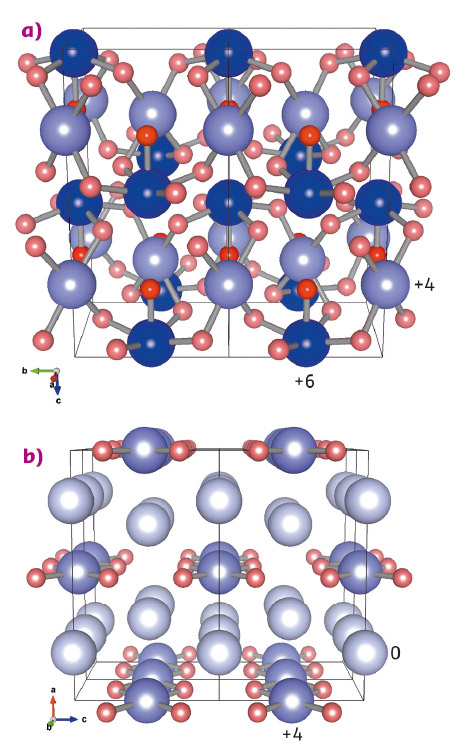
From different starting mixtures, two oxides, Xe2O5 and Xe3O2, have been synthesised and their structures, which were predicted to have the lowest enthalpies by ab initio modelling, are represented in Figure 136. Xenon atoms adopt mixed oxidation states of 0 and +4 in Xe3O2 and +4 and +6 in Xe2O5. Both oxides form extended networks incorporating oxygen-sharing XeO4 squares, while Xe2O5 additionally incorporates oxygen-sharing XeO5 pyramids.
 |
|
Fig. 136: Structures of the stable xenon oxides at 82 GPa. a) Xe2O5 and b) Xe3O2. Xenon atoms are shown in blue and oxygen atoms in red. The oxygen atoms have an oxidation state of –2, and the darker shade of red indicates an oxygen atom that bonds with only one xenon atom. The oxidation states of the xenon atoms are indicated by different shades of blue. |
The reactivity of xenon and oxygen is predicted to further increase with pressure, yielding new oxides such as XeO2 and XeO3. It is interesting to note that xenon atoms adopt mixed valence states in the oxides stable at the lowest pressure, i.e. compounds with unusual stoichiometries. This may be a general trend for compounds formed under high compression. Xenon is also predicted to react with other elements that are abundant in the earth, such as iron and nickel [1]; this remains to be confirmed experimentally. Although the laboratory syntheses of Xe2O5 and Xe3O2 don't directly solve the missing Xe problem — the element's scarcity makes it unlikely to form its own separate phase in the mantle — they demonstrate that this element is even more reactive under pressure than previously predicted.
Principal publication and authors
Synthesis and stability of xenon oxides Xe2O5 and Xe3O2 under pressure, A. Dewaele (a), N. Worth (b), C.J. Pickard (c,d,e), R.J. Needs (b), S. Pascarelli (f), O. Mathon (f), M. Mezouar (f) and T. Irifune (g,h), Nature Chem. 8, 784–790 (2016); doi: 10.1038/nchem.2528.
(a) CEA, Direction des applications militaires, Arpajon (France)
(b) Cavendish Laboratory, University of Cambridge (UK)
(c) Department of Physics and Astronomy, University College London (UK)
(d) Department of Materials Science & Metallurgy, University of Cambridge (UK)
(e) Advanced Institute for Materials Research, Tohoku University, Sendai (Japan)
(f) ESRF
(g) Ehime University, Matsuyama (Japan)
(h) Earth-Life Science Institute, Tokyo Institute of Technology (Japan)
References
[1] L. Zhu et al., Nature Chem. 6, 644 (2014)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.