- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2015
- Electronic structure, magnetism and dynamics
- Combining hard and soft XAS to characterise the deactivation pathway of an immobilised transfer hydrogenation catalyst
Combining hard and soft XAS to characterise the deactivation pathway of an immobilised transfer hydrogenation catalyst
A multi-element X-ray absorption spectroscopy study was performed on an immobilised iridium transfer hydrogenation catalyst. Ex situ measurements probing the Ir L-edge and the Cl-K edge showed the loss of an Ir-Cl bond during activation of the catalyst, followed by further loss of a second Ir-Cl bond throughout the depreciation of catalytic activity. Build-up of potassium cation, from the KOtBu used as co-catalyst, was detected in the deactivated samples. An operando experiment was carried out using a spectroscopic flow cell to monitor the loss of chloride and accretion of potassium throughout deactivation. Its findings corroborated the ex situ measurements and kinetic data to support a second order deactivation mechanism.
Immobilisation of molecular catalysts is a common strategy in modern synthetic science to facilitate catalyst recovery and reuse. This allows reduction of the cost of application of these often complex and sophisticated catalysts while improving the sustainability of such manufacturing processes, particularly when precious metals are used. Most immobilisation technology, however, is developed empirically due to a lack of suitable and effective characterisation techniques once the catalyst is immobilised. It is quite common to find the behaviour of the catalyst changed upon immobilisation through either interaction with the solid support or isolation from other catalyst molecules.
We have previously reported a novel approach to immobilisation of organometallic catalysts containing the Cp* ligand [1]. By linking the catalyst, [Cp*IrCl2]2, with the support through the strongly binding Cp* ligand, metal leaching is suppressed to a negligible level. Slow deactivation over 30 recycling uses was however still observed due to chemical changes at the metal centre. Thus, an XAS study was developed to characterise the catalyst deactivation process.
A novel multi-element approach was adopted to fully characterise the catalyst at various stages of the deactivation process. Instead of solely observing the Ir centre through Ir L-edge (11.2 keV), both the Ir L-edge EXAFS and Cl K-edge XANES were probed, using beamline BM28 (XMaS CRG). Ex situ spectra of the fresh catalyst, and catalysts after 3 (still highly active) and 30 cycles of use (inactive) clearly showed a rapid loss of about 50% of the Cl and Ir-Cl signals, without loss of catalytic activity, followed by a slow loss of the remaining chloride ligand and subsequent deactivation. These results link the deactivation process with a second ligand exchange of the immobilised catalyst.
Treatment of the deactivated catalyst with dilute aqueous hydrochloric acid temporarily restored catalytic activity, which was rapidly lost after 3 cycles of use. Cl XANES spectrum of this ‘reactivated‘ catalyst indicated a change in speciation of the Cl content to KCl. This new and unexpected observation pointed to a presence in the deactivated catalyst of the potassium cation, introduced in the form of co-catalyst KOtBu under catalytic conditions. A hypothesis on the activation and deactivation of this immobilised catalyst, taking into account all experimental observations, was developed (Figure 72).
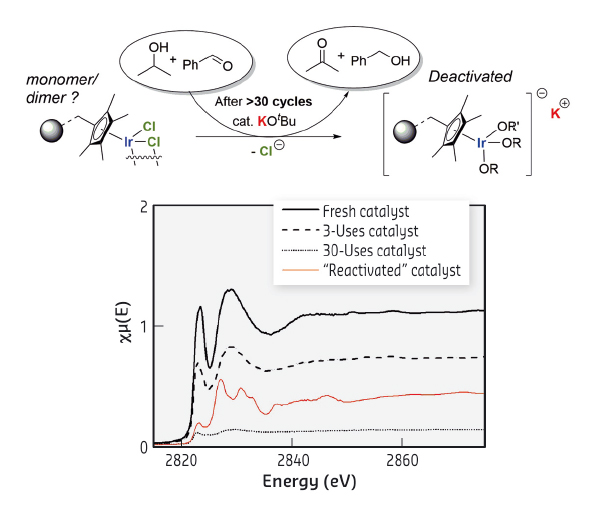 |
|
Fig. 72: Proposed deactivation form of the immobilised catalyst and Cl K-edge XANES spectra of the catalyst at various stages. |
To correlate the structural changes above with changes in catalytic activity, an operando experiment was performed using a spectroscopic flow-cell. Spectra of the catalyst at Cl K-edge and K K-edge were monitored over 24 hours as the reaction mixture was flowed through the catalyst. The decrease in catalytic activity was found to match the concurrent decrease of Cl content and increase of K content, confirming the hypothesis above (Figure 73).
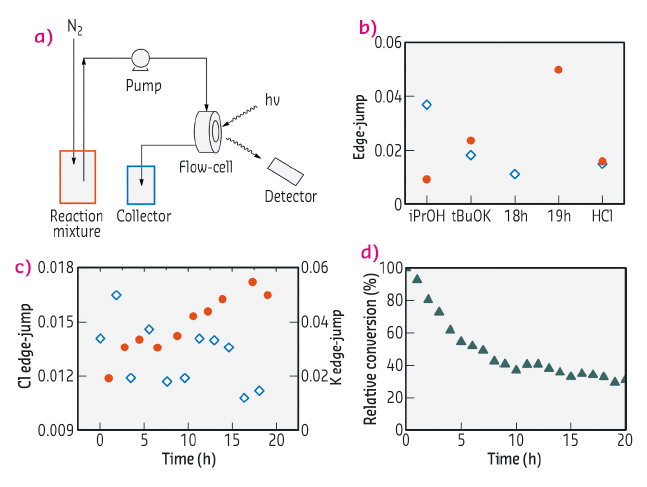 |
|
Fig. 73: (a) Operando experimental setup; (b) Cl (blue) and K (red) edge jumps at various stages of the experiment; (c) Cl (blue) and K (red) edge jumps during catalytic reaction; (d) concurrent catalytic activity vs time. |
The results, particularly the unexpected detection of the potassium cation in the deactivated catalyst, highlight multi-element XAS as a powerful combination of spectroscopic techniques for immobilised molecular catalysts. The specific deactivation pathway determined in this study will now be pivotal in developing strategies to reactivate the catalyst or to suppress catalyst deactivation.
Principal publication and authors
Activation and deactivation of a robust immobilized Cp*Ir-transfer hydrogenation catalyst: a multi-element in situ X-ray absorption spectroscopy study, G.J. Sherborne (a), M.R. Chapman (a), A.J. Blacker (a), R.A. Bourne (a), T. Chamberlain (b), B.D. Crossley (c), S.J. Lucas (a), P.G. McGowan (a), M.A. Newton (d), T.E.O. Screen (c), P. Thompson (d), C.E. Willans (a) and B.N. Nguyen (a), JACS 137, 4151-4157 (2015); doi: 10.1021/ja512868a.
(a) Institute of Process Research and Development, School of Chemistry, University of Leeds (UK)
(b) School of Chemistry, University of Nottingham (UK)
(c) Yorkshire Process Technology Ltd., Leeds Innovation Centre, Leeds (UK)
(d) Department of Physics, University of Liverpool (UK); Xmas CRG, ESRF, Grenoble (France)
References
[1] S.J. Lucas et al., Chem. Commun. 49, 5562-5564 (2013).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.