Methods for structural biology
Development of Powder Diffraction Methods for Structural Biology
We have been developing the application of powder diffraction to proteins, using, in particular, high resolution powder diffraction on ID31, but also exploiting the very high detector efficiency of area detectors on ID11, with occasional measurements on BM01A and BM16 (SAXS). The work has been the basis for three successful projects for students in chemistry from Bath university spending a year at ESRF (S. Basso, 2004–5, M. Jenner, 2005–6, L. Knight, 2006–7) and two new projects with S. Dagogo (2007–8) and Yves Watier (PhD candidate: 2007–2010).
Results to date include structure determination via the molecular replacement method and structure refinement via combined Rietveld and stereochemically restrained analysis [1–3], model building accomplished by difference Fourier and OMIT methods [3], extraction of solvent envelopes [4, 5], protein cryocooling [6], improved methods for data collection [7] and interpretation [8], and high-throughput phase identification in the cases of lysozyme [2] and insulin from different sources, the latter being Lisa Knight's project during her year in Grenoble.
We have shown that structure refinements can be improved by the use of multiple datasets, provided there are no significant changes in structure factors between datasets. Two independent investigations of Hen Egg White Lysozyme (HEWL) indicated that the use of multiple patterns associated with slightly different lattice parameters leads to enhanced extracted intensities and more robust structure refinements [2, 9]. We prepared a total of 44 different polycrystalline HEWL precipitates at 277 K and at room temperature and in the pH range between 6.56 and 3.33 [2]. High resolution powder diffraction data were collected at room temperature (λ= 1.249826 Å on ID31). The anisotropic effect of pH of crystallisation on the lattice of tetragonal HEWL shifts the peaks significantly and alleviates the peak overlap problem. Figure 1 (lower left) shows the significant improvement of the effective completeness as a result of the use of multiple profiles during intensity extraction. A more detailed description of this refinement can be found elsewhere [2] and the model is deposited in the pdb database with access code 2A6U. The upper panels of figure 1 show the excellent Rietveld fit which could be achieved. We found that this approach resulted in a much smoother and more robust refinement than previously experienced with single pattern fits [1] and produced a structural model with excellent stereochemistry (the resulting Ramachandran plot is shown on the lower right panel of figure 1).
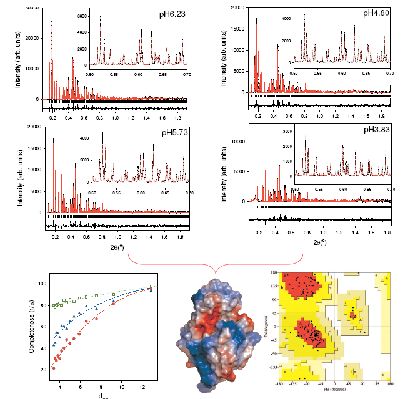
Figure 1: Upper Panels: The multi-data-set Rietveld refinement for HEWL samples at pH6.23, pH5.73, pH4.80 and pH3.83 and crystallized at 277 K. Lower-left: Effective completeness for the powder diffraction data at a 1σ level where red circles represent a single powder pattern, blue triangles are the combined fit to four patterns and the open squares show the maximum completeness attainable in this case. The dotted lines are guides to the eye. Lower-middle: The refined conformation of tetragonal HEWL, in total 7 helices and 3 strands were observed. Lower-right: Ramachandran plot for main-chain torsion angles (φ, ψ). 94 out of the 113 non-proline, non-glycine residues are in the most favoured regions (A,B,L - red) [2].
In one of our most recent studies of the second SH3 domain of ponsin [3], we exploited radiation induced anisotropic lattice strains in a specially modified multi-pattern Pawley refinement taking into consideration likelihood criteria for partitioning overlapped peaks [8]. The powder extracted data were sufficient for the structure solution of the domain via the molecular replacement method (figure 2). Maximum likelihood refinement improved the phases to a level where we could trace the main chain alterations, build additional residues where needed and eventually place the correct side chains along the sequence; a substantial result from powder diffraction data. The protein conformation was refined in a multiple data set stereochemically restrained Rietveld analysis taking advantage of sample and radiation-induced anisotropic lattice strains. We further benefited from an approach combining multiple-data-set Rietveld analysis and periodical OMIT map [10] computation, to reduce the bias of the final model, extending the resolution limits to levels comparable to single-crystal measurements, and even detecting several water molecules bound to the protein [3]. The benefits from the multi-pattern approach are perhaps most striking from the visual inspection of the total OMIT map computed at the final steps of analysis shown in figure 2.
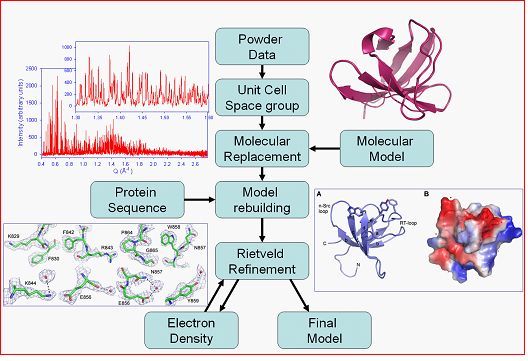
Figure 2: Powder diffraction data analysis procedure followed for structure solution via the molecular replacement method, model building and structure refinement. The data and model shown correspond to the second SH3 domain of ponsin [3] and final omit maps are shown on the lower left.
When there is no starting model available for a protein molecule then we are faced with the crystallographic phase problem. The isomorphous replacement method [11] determines the phases of X-ray reflections by comparing data from a series of crystals which contain different additive heavy atoms. In the case of a Gadolinium derivative of hen egg white lysozyme [4] we prepared derivatives using different concentrations of the Gd and also different pH values in order to alleviate the peak overlap problem. Extracted intensities were then used to solve for the atomic positions of Gd using Shelxd [12] and then refinement of the heavy atom substructure and phasing were carried out using Sharp [13]. Figure 3 shows the solvent channels in the protein structure determined during this phasing procedure and the positions of the molecules from the known structure [4].
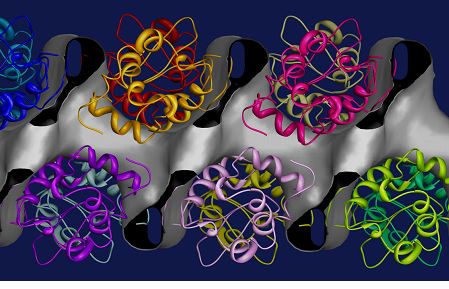
Figure 3: Solvent channels in Hen Egg White Lysozyme crystals. The molecular envelope derived via single isomorphous replacement using a Gadolinium derivative is represented as gray surface [4]. the figure shows the linear solvent channel which traverses the crystal parallel to the c axis (horizontal display direction). The protein crystal structure, represented as a main-chain ribbon model, is superimposed on this map.
In order to achieve the ambitious goals of the current project, we have assembled an international team of research associates with complementary backgrounds and specializations, including university departments (e.g. EMBL, Hamburg, EPFL, Lausanne, and the Institute of Crystallography, Bari, Italy), companies (Panalytical in The Netherlands - interested in the diffraction aspects - and diabetes-care company Novo Nordisk in Denmark) and central facilities (APS, SLS, ISIS, Spring-8).
Following the award of beam time for a three-year long-term project at the ESRF to Marc Schiltz (EPF Lausanne) and our group (ESRF), and our results to date, it is clear that work carried out at ESRF preponderates in this expanding area, exemplified by the award of the 2006 EPDIC prize to Irene for her work on protein powder diffraction. She has recently written an invited review article for the special issue of Acta Crystallographica (60th anniversary of IUCr) [14] and will be a keynote lecturer at the 2008 IUCr congress in Osaka. Finally, an international workshop on "Development and Directions of Powder Diffraction on Proteins" was recently organized at the ESRF on 22–23 June 2007, with support from the International Union of Crystallography, the British Crystallographic Association, Panalytical and the EPFL. This event will be followed by a second meeting which will be held in Kyoto as a satellite workshop of the IUCR congress.
References:
[1] Margiolaki et al . (2005). Acta Cryst. D61. 423-432 . See also: ESRF Scientific Highlights 2004, p. 30-31.
[2] Basso, S. et al. (2005). Acta Cryst. D61, 1612-25, doi:10.1107/S0907444905031963
[3] Margiolaki et al. (2007). J. Am. Chem. Soc. ASAP Article 10.1021/ja073846c S0002-7863(07)03846-2 (Web Release Date: September 5, 2007)
[4] Wright et al. (2007). J. Appl. Cryst. (Submitted); See also: ESRF Scientific Highlights 2006, p. 61-62.
[5] Besnard et al. (2007). Z. Kristallogr. (In press)
[6] Jenner et al. (2007). J. Appl. Cryst. 40, 121-124.
[7] Margiolaki et al. (2007). Z. Kristallogr. (In press)
[8] Wright et al. (2007). Z. Kristallogr. (In press)
[9] Von Dreele, R. B. (2007) J. Appl. Cryst. 40 133-143.
[10] Bhat, T. N. (1988) J. Appl. Cryst. 21, 279-281.
[11] Perutz, M. F., (1956) Acta Cryst. 9, 867-873.
[12] Uson, I. and Sheldrick, G. M., (1999) Curr. Op. Struct. Biol., 9 643-648.
[13] La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol. 276, 472-494.
[14] I. Margiolaki & J. P. Wright, Acta Crystallogr A. 2008 Jan 64(Pt 1):169-80.
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.