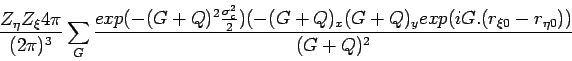| (1) |
and
| (2) |
| (3) |
where
![]() and
and ![]() is the charge of the
is the charge of the
![]() ion.
ion.
| (4) |
| (5) |
| (6) |
Alessandro Mirone 1 and Matteo d'Astuto
March 21, 2006
The code is written in Python using its numerialc library NumPy. In order to use the program just a very basic knowledge of the language is required. One-half-a-day tutorial is available at www.python.org.
Lattice Hamiltonian is developed to second order around the nominal equilibrium position, given by the experimental crystal structure. This corresponds to a simple implementation of the quasi-harmonic approximation (the positions of the atom at rest are choosen at the minimum of the free energy, in thermodynamic equilibrium) instead of the harmonic approximation (the position of the atom at rest are choose at the minimum of the potential energy, in mechanical equilibrium)
The Hamiltonian is given by the sum of the kinetic part plus the sum of interatomic two-body central potentials. The Hamiltonian degrees of freedom are the atomic cores and atomic shells coordinates. The atomic shells have zero mass and does not contribute to the kinetic part.
The potentials implemented up to now in OpenPhonon are:
| (1) |
and
| (2) |
| (3) |
where
![]() and
and ![]() is the charge of the
is the charge of the
![]() ion.
ion.
| (4) |
| (5) |
| (6) |
Finally, in condensed matter (in particular for metals)
the Coulomb potential is renormalized
by the screening effect of the surrounding charges.
This effect can be calculated in many ways,
we have implemented the simplest approach: the
Thomas-Fermi one, i.e.
replacing the Coulomb potential by the
Yukawa potential:
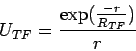 |
(7) |
The motion of the atom labelled ![]() in the
in the ![]() th cell is described by the following equations:
th cell is described by the following equations:
where equation (8) describes the motion of the centre of mass
of each atom while equation (9) refers to the motion of the
shell associated to each atom, and its mass
![]() .
The parameter
.
The parameter ![]() is given by
is given by
![]() are the spring constants which tie the shells to the cores. Symbols
are the spring constants which tie the shells to the cores. Symbols
![]() ,
,![]() ,
,![]() ,
,![]() denote non-coulombian interaction.
The
denote non-coulombian interaction.
The ![]() represent interactions between the cores.
Although coulombian interactions act between the cores too,
they need a special treatment because of their long range, and
have thus their own writing
represent interactions between the cores.
Although coulombian interactions act between the cores too,
they need a special treatment because of their long range, and
have thus their own writing ![]() .
.
![]() are the elastic interactions between a shell and a core on another
site,
are the elastic interactions between a shell and a core on another
site, ![]() between a core and another site shell and
between a core and another site shell and ![]() are the
interactions between two different shells.
These terms are the sum of analytical potentials ( Born-Mayer or Van de Waals )
and Born-von-Karman potentials.
In the case of analytical potentials the
are the
interactions between two different shells.
These terms are the sum of analytical potentials ( Born-Mayer or Van de Waals )
and Born-von-Karman potentials.
In the case of analytical potentials the ![]() 's are derived from the
analogous of equation (10) while in the Born-von-Karman
case they are derived from the longitudinal and transverse second
derivatives
's are derived from the
analogous of equation (10) while in the Born-von-Karman
case they are derived from the longitudinal and transverse second
derivatives ![]() and
and ![]() , namely :
, namely :
| (11) |
![]() being the unit vector going from
being the unit vector going from
![]() to
to
![]()
One can cast together
![]() (core charge) and
(core charge) and
![]() (shell charge) in equations
(8) and (9) to get
(shell charge) in equations
(8) and (9) to get
![]() (ion charge=core+shell) and
(ion charge=core+shell) and
![]() by using a different set of displacement variables (
by using a different set of displacement variables (
![]() and
and
![]() ) where
) where
![]() .
By performing such substitution, by summing together equations
(8) and (9) and considering the limit
.
By performing such substitution, by summing together equations
(8) and (9) and considering the limit
![]() , one obtains :
, one obtains :
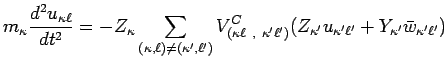 |
|||
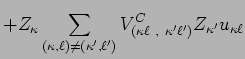 |
|||
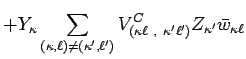 |
(12) | ||
![$\displaystyle - \sum_{(\kappa,\ell)\neq(\kappa',\ell')}\Bigl[
V_{(\kappa \ell ~...
...ll ~,~\kappa ' \ell ' )}^{T'}
\Bigl] (u_{\kappa ' \ell ' } - u_{\kappa \ell } )$](img48.png) |
|||
![$\displaystyle - \sum_{(\kappa,\ell)\neq(\kappa',\ell')}\Bigl[
V_{(\kappa \ell ~...
...S} +
V_{(\kappa \ell ~,~\kappa ' \ell ' )}^{T}
\Bigl] \bar w_{\kappa ' \ell ' }$](img49.png) |
|||
![$\displaystyle + \sum_{(\kappa,\ell)\neq(\kappa',\ell')}\Bigl[
V_{(\kappa \ell ~...
...}^{S} +
V_{(\kappa \ell ~,~\kappa ' \ell ' )}^{T'}
\Bigl] \bar w_{\kappa \ell }$](img50.png) |
and for the zero-mass shells
Phonon eigenvectors can be written as Bloch waves:
where ![]() and
and ![]() are now
are now ![]() dimensional vectors ( N being the number of atoms in a cell),
Y,Z,K are
dimensional vectors ( N being the number of atoms in a cell),
Y,Z,K are ![]() dimensional diagonal matrices whose diagonals are equal to the shell charges, ionic charge and
spring constants respectively. The matrices C (coulombian), and the V's are obtained substituting equation 14
into 15 and summing over all the cells up to convergence. The subscript ``
dimensional diagonal matrices whose diagonals are equal to the shell charges, ionic charge and
spring constants respectively. The matrices C (coulombian), and the V's are obtained substituting equation 14
into 15 and summing over all the cells up to convergence. The subscript ``![]() '' in
'' in ![]() and in
and in ![]() 's denotes the so said ``self-forces''.
The dynamical matrix is found from equation (15) by elimination of
's denotes the so said ``self-forces''.
The dynamical matrix is found from equation (15) by elimination of ![]() .
The summation of
.
The summation of ![]() and
and ![]() over the cells needs a special treatment because of the Coulomb
long range.
over the cells needs a special treatment because of the Coulomb
long range.
Coulomb long-range makes the real-space summation of forces very slow because of the
![]() behaviour. On the other hand in reciprocal spaces Colomb interaction
behaves as
behaviour. On the other hand in reciprocal spaces Colomb interaction
behaves as ![]() for a point charge. Convergence at high
for a point charge. Convergence at high ![]() can be accelerated
by gaussian-spreading the point charges, and introducing thus a gaussian factor in the Coulomb interaction.
This spreading changes the dynamical matrix, by a little amount if the spreading is small, and
the correct result can be recovered by subtracting the difference between the field of a gaussian charge
and the field of a point charge. This difference tends to zero very rapidly for distances bigger than
the charge spreading and thus the real-space summation converges vary fast.
The Coulomb contribution to the dynamical matric is thus made of two contribution :
a reciprocal space summation for spreaded charges, and a real space summation for the difference between
a point charges and the spreaded charges.
can be accelerated
by gaussian-spreading the point charges, and introducing thus a gaussian factor in the Coulomb interaction.
This spreading changes the dynamical matrix, by a little amount if the spreading is small, and
the correct result can be recovered by subtracting the difference between the field of a gaussian charge
and the field of a point charge. This difference tends to zero very rapidly for distances bigger than
the charge spreading and thus the real-space summation converges vary fast.
The Coulomb contribution to the dynamical matric is thus made of two contribution :
a reciprocal space summation for spreaded charges, and a real space summation for the difference between
a point charges and the spreaded charges.
A point charge ![]() is spreaded as
is spreaded as ![]() :
:
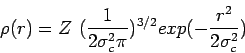
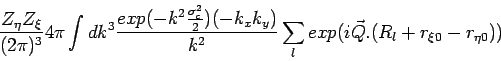
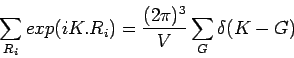
while the self term (![]() of equation(15)) for atom
of equation(15)) for atom ![]() is obtained by setting
is obtained by setting ![]() and
and ![]() in the above expression.
In calculating the term given by formula (16) we are actually doing an error : as we
sum over the whole lattice we are considering also a spurious affect coming from the interaction of the charge
with itself. In a case of rigid ions the self term
in the above expression.
In calculating the term given by formula (16) we are actually doing an error : as we
sum over the whole lattice we are considering also a spurious affect coming from the interaction of the charge
with itself. In a case of rigid ions the self term ![]() simply subtracts to
simply subtracts to ![]() and the spurious
terms in
and the spurious
terms in ![]() and in
and in ![]() would delete each other. However in the case of polarizable shells, as can be seen
in equation (15),
would delete each other. However in the case of polarizable shells, as can be seen
in equation (15), ![]() and
and ![]() have a life on their own, being multiplied sometimes by
have a life on their own, being multiplied sometimes by ![]() and some other time by
and some other time by ![]() . The spurious term has thus to be removed from
. The spurious term has thus to be removed from ![]() and
and ![]() . That can be obtained
removing to the diagonal blocks of
. That can be obtained
removing to the diagonal blocks of ![]() and
and ![]() , for each atom
, for each atom ![]() the following quantity :
the following quantity :
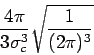
The following subsection explains how the difference between a point charge and a gaussian one is recovered
The first step is to run the program preparation.py, followed by the name of a file containing the structure :
python preparation.py <structure_file_name>( see section 6 to see how to invoke OpenPhonon at ESRF or with the pre-compiled version). The structure file specifies the atoms and their position in the cell. This is accomplished by setting the variables AtomNames, CellVectors, PositionsList at the beginning of the input file ( note that the input file is simply executed as a Python script, so one has to use Python sintax).
We show here an example input file for preparation.py referring to a literature case [4]. It can be found in the distribution archive ( binary or source) in the directory test_OP:
AtomNames=['Cu', 'O', 'Nd' ]
cellvectors=Numeric.array(
[
[3.95 , 0.0 , 0.0],
[0.0 , 3.95 , 0.0],
[0.0 , 0.0 , 12.06556]
] )
PositionsList=[
Numeric.array(
[ [0.0, 0.0, 0.0 ],
[0.5, 0.5, 0.5 ]
]
),
Numeric.array(
[ [0.5, 0.0, 0.0 ],
[0.0, 0.5, 0.0 ],
[0.5, 0.0, 0.25 ],
[0.0, 0.5, 0.25 ],
[1.0, 0.5, 0.5 ],
[0.5, 1.0, 0.5 ],
[1.0, 0.5, 0.75],
[0.5, 1.0, 0.75],
]
),
Numeric.array(
[ [0.0, 0.0, 0.352],
[0.5, 0.5, 0.148],
[0.5, 0.5, 0.852],
[1.0, 1.0, 0.648]
]
)
]
SeekedTetragon=[('Cu',0),('O',1),('Nd',0),('Nd',2)]
CellsCoo=[(0,0,0),(0,0,0),(0,0,0),(0,0,0)] # optional
dr_shell=0.05 # optional
num_nei=3 # optional
The SeekedTetragon array must specify a non degenerate 3-D solid that is rotated
and translated in all possible ways to look for symmetries and equivalent atoms.
CellsCoo defines the cell in which the vertices are. This variable is optional and is set by default to all vertices in the same cell.
dr_shell defines the width of a shell. Two-body bonds are catalogued according to their length and the type of atoms that they concern. This variable is set by default to 0.1.
Finally num_nei, which is set by default to 2, is the number of neighbours.
The distribution archive contains an example file ( preparationinput ).
This file being set, the script preparation.py can be run (python preparation.py structure-file-name). The script does a rough evaluation of the symmetries and of inequivalent bonds and creates two files. Both files need to be stored somewhere for subsequent use by the program that calculates dispersions. One of these file, cella contains all the needed structural informations, while the other,parinput is a template file that can be edited with the parameters of the interatomic potentials as explained in the next subsection.
The distribution archive contains parinputedited that corresponds to the test case [4]. Such a file describes the interatomic potentials. Units are CGS. Some knowledge of Python and of the Numerical extension (NumPy) is necessary. Interactions are book-keept by a Python dictionary. For example the dictionary entry
BK_L['O_G1']['Nd_G0']describes the longitudinal Born-Karman interactions between oxygen atoms of the equivalency group
# XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX # Interactions between 'O_G1' and 'Nd_G0' # Shell N0 going from 2.327062 to 2.327062 like O7 in 0 and Nd2 in 0 0 0 # Shell N1 going from 4.584508 to 4.584508 like O7 in 0 and Nd2 in -1 0 0 # Shell N2 going from 5.192371 to 5.192371 like O7 in 0 and Nd1 in 0 0 1 BMpar.A= 2000 * ev_erg BMpar.R= 0.316 * A_cm distances= [ 2.327062 * A_cm ,4.584508* A_cm ] BMpar.ZZ= Z_['Nd_G0']*Z_['O_G0' ] print " ['Nd_G0']*Z_['O_G0' ] " BK_L['O_G1']['Nd_G0']=map(bm_L,distances) BK_T['O_G1']['Nd_G0']=map(bm_T,distances)
where ![]() is an user defined function (see input file)
of distance.
is an user defined function (see input file)
of distance.
The parameters file specifies also other thing like, atom polarizability, Charges and analytical potentials. A few words should be spent on analytical potentials that can be of the Born-Mayer, Van der Walls, Lenard Jones type. They are specified by atom-dependent parameters, contrarily to the spring constants which are interaction (bond) dependent, .i.e. are given for each couple of interacting atom-types.
However in the literature one often finds analytical parameter that are bond dependent.
In this case on can include them as spring constant. this is done in our example perinputedited file,
where the function ![]() is mapped on the shell distances to get spring
constants.
is mapped on the shell distances to get spring
constants.
Then the calculation is performed running dispersionDeb.py under Python with three arguments:
python dispersionDeb.py <parinputedited-filename> <cella-filename> <additional-filename>
The significance of parinputedited and cella has been explained before. The last parameter specifies a file containing some parameters concerning the q-points at which phonons are calculated and other parameters governing the treatement of coulomb interactions. This is an examples :
astar=2*math.pi/3.95 cstar=2*math.pi/12.06556 eps=0.0011 Q=Numeric.array( [ [astar*0.01*i+eps,-astar*0.01*i+eps,0] \ for i in range(1,100)]) debyerange=[8,8,2] debye=astar/4 SMcellrange=[2,2,2] cellrange= [-1,-1,-1] Kcellrange=[8,8,2] sigmacellrange=[8,8,2] sigmacharge=3 IS_IT_SCREENED=1 selectors=[(1.0,1.0,0.0), (1.0,-1.0,0.0), (1.0,0.0,1.0), (-1.0,1.0,0.0)]
The distribution archive contains an examples file (dispersionDebinput).
The q-points are defined by the variable Q, cstar and astar being just auxiliary variables.
When summing up the contributions from spring constants a loop is done over all atoms in several cells. The variable SMcellrange gives the extent of this loop over the unit cells
in the three directions a,b,c. Giving
![]() , for example,
implies considering just the central unit-cell, but that might loose several interactions,
particularly in the considered case where we include also second neighbours.
For the example considered,
, for example,
implies considering just the central unit-cell, but that might loose several interactions,
particularly in the considered case where we include also second neighbours.
For the example considered,
![]() is enough.
The variable
is enough.
The variable ![]() specifies the extent of the summation of analytical potentials,
namely Born-Mayer, Van der Waals, Lennard Jones.
The variables
specifies the extent of the summation of analytical potentials,
namely Born-Mayer, Van der Waals, Lennard Jones.
The variables
![]() determines if the Coulomb interactions are screened or not.
In the case of unscreened interactions the summation shows convergence problems
that are cured by decomposing it partly in reciprocal space
and partly in real space thanks to an appropriate charge smearing ( see Unisoft manual
for details [5]). The Gaussian charge smearing is governed by
determines if the Coulomb interactions are screened or not.
In the case of unscreened interactions the summation shows convergence problems
that are cured by decomposing it partly in reciprocal space
and partly in real space thanks to an appropriate charge smearing ( see Unisoft manual
for details [5]). The Gaussian charge smearing is governed by ![]() , given in
Angstroem.
The extensions of reciprocal-space and real-space summation are given by the
variables
, given in
Angstroem.
The extensions of reciprocal-space and real-space summation are given by the
variables ![]() and
and
![]() respectively. Taking a large sigma for charge smearing will make Kcellrange smaller( faster reciprocal space convergence) but will give a bigger sigmacellrange.
Convergence has to be checked against these variables.
respectively. Taking a large sigma for charge smearing will make Kcellrange smaller( faster reciprocal space convergence) but will give a bigger sigmacellrange.
Convergence has to be checked against these variables.
In the case of a screened potential, two variables have to be given : ![]() which multiplies
the distance in the exponential damping of the Coulomb potential and the extent of the real space
summation, debyerange.
which multiplies
the distance in the exponential damping of the Coulomb potential and the extent of the real space
summation, debyerange.
Finally the variable ![]() gives a set of directions
against which the simmetry of the eigenmodes is checked.
gives a set of directions
against which the simmetry of the eigenmodes is checked.
The output is contained in three files : storeall, disp_res, disp_ressq. The ASCII files disp_res, disp_ressq contain the squares and the values of the phonon frequencies respectively. The file storeall stores in Python ``pickled'' form the calculated eigenvectors and frequencies. This file is finally used by the program scattering_shifted.py to calculate scattering intensities.
If the variable ![]() is set, an additional file, named polar_res,
where for each directions the index of the eigenvalues ( referring to the
ordering given in the output file) is given following the order of growing eigenvector
projections over the
is set, an additional file, named polar_res,
where for each directions the index of the eigenvalues ( referring to the
ordering given in the output file) is given following the order of growing eigenvector
projections over the ![]() direction. This can help to retrieve
eigenvectors of specific symmetries, whose signal is enhanced by
the experimental geometry.
direction. This can help to retrieve
eigenvectors of specific symmetries, whose signal is enhanced by
the experimental geometry.
The general expression for the dynamical matrix for the dipolar fluctuation model
(DFM) is [8]:
USE_DFM=1
Then one has to fill in the entries for Tens_P, Tens_S, Tens_T.
Tensor K is scaled to ![]() by rescaling S and T [8].
The following is an example for hcp Cobalt :
by rescaling S and T [8].
The following is an example for hcp Cobalt :
# XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
# Tensorial Interactions between 'Co_G0' and 'Co_G0'
# Shell N0 going from 0.996332 to 0.996332 like Co1 in 0 and Co0 in 1 0 1
# Shell N1 going from 1.000000 to 1.000000 like Co1 in 0 and Co1 in -1 1 0
# Shell N2 going from 1.411622 to 1.411622 like Co1 in 0 and Co0 in -1 1 0
# Shell N3 going from 1.624000 to 1.624000 like Co1 in 0 and Co1 in 0 0 1
# Shell N4 going from 1.729936 to 1.729936 like Co1 in 0 and Co0 in 0 2 1
# Shell N5 going from 1.732051 to 1.732051 like Co1 in 0 and Co1 in -2 1 0
# Shell N6 going from 1.907191 to 1.907191 like Co1 in 0 and Co1 in 0 1 -1
# Shell N7 going from 2.000000 to 2.000000 like Co1 in 0 and Co1 in 0 2 0
hcp_a=2.51
hcp_c=1.624*hcp_a
(a1,b1,g1,d1,
a2,e2,b2,g2,
a3,b3,g3,d3)= [0]*12
(a1,b1,g1,
a2,b2,g2,e2,
a3,b3,g3)= ( 11925.5,
-1267.4,
36831.4,
-1819.8,
40089.8,
2171.9,
1502.5,
779.8,
455.2,
-3507.4)
Tens_P['Co_G0']['Co_G0']=[ [Numeric.array([ hcp_a/math.sqrt(3.0) ,
0, hcp_c/2.0 ] ),
Numeric.array([[a1,0 ,d1],
[0 ,b1,0 ],
[d1,0 ,g1]])
],
[Numeric.array([ 0 , hcp_a , 0 ] ),
Numeric.array([[a2 ,e2 ,0],
[-e2 ,b2 ,0 ],
[ 0 ,0 ,g2 ]])
],
[Numeric.array([ -2*hcp_a/math.sqrt(3.0) ,
0, hcp_c/2.0 ] ),
Numeric.array([[a3 ,0 ,d3],
[0 ,b3 ,0 ],
[d3 ,0 ,g3 ]])
]
]
one can see that the general form of Tens_P is given, for each shell, by a typical site-to-site vector and the corresponding tensor. The other tensors for the other interaction of the same shell are found by similitude transforming according to the simmetry group of the crystal. Tens_S and Tens_T are specified in analogous way.
Jahn-Teller tensorial forces can be treated in an analogous way[6,7].
The parameters Tens_JT has to be specified in the same way as
Tens_P, Tens_S, Tens_T but in addition one has to add for each interaction
two vectors that are used to calculated the JT factor: this factor is given by the product
of two sines having as arguments the scalar product of ![]() and the vectors.
The JT model is activated by setting the variable USE_JT in the input file for dispersionDeb.py :
and the vectors.
The JT model is activated by setting the variable USE_JT in the input file for dispersionDeb.py :
USE_JT=1the way to input the vectors for the JT factor is shown as follows :
B_JT=100000.0
a=3.95
b=a
Tens_JT={}
Tens_JT['O_G0'] = {}
Tens_JT ['O_G0']['O_G0']= [
[
Numeric.array([a/2.0,b/2.0,0]),
Numeric.array([[0,B_JT,0.0],
[0,0,0.0],
[0,0,0.0]] ),
Numeric.array([a/2.0,0,0]),
Numeric.array([0,b/2.0,0])
],
[
Numeric.array([a,0,0]),
Numeric.array([[-B_JT,0.0,0.0],
[0,0,0.0],
[0,0,0.0]] ),
Numeric.array([a/2.0,0,0]),
Numeric.array([a/2.0,0,0])
]
]
where the vectors follows the ![]() tensor.
tensor.
The following input example ( file parinputedited )
angleBonds={}
angleBonds['Sb_G0']={}
angleBonds['Sb_G0']['Sb_G0']={}
angleBonds['Sb_G0']['Sb_G0']['Co_G0']=[
[
Numeric.array([0.25 , 0.09212, -0.08537])*9.035,
0.0002 ,
Numeric.array([0. , -2*0.15788 , 0.])*9.035,
0.0002,
anglestrenght,
]
establishes an angular spring for the angle having vertex Sb_G0 ( the first key of angleBonds ) and two sides , one made by the segment connecting Sb_G0 to Sb_G0 (second key) and having components dx,dy,dz = Numeric.array([0.25 , 0.09212, -0.08537])*9.035 . The other side goes from the vertex to Co_G0 and has components Numeric.array([0. , -2*0.15788 , 0.])*9.035.
To activate these interaction you have to write the following keywoards in the input file
USE_ANGLE_BOND=1 AB_cellrange=[1,1,1]
A scan for every possible atomic triplet is done looping over a cell range whose span is given by the three components AB_cellrange=[1,1,1]. All rotations of the structure symmetry group are then applied to every triplet to check if a rotation can bring the triplet to coincide with the one given by angleBonds, with a tolerance given by 0.0002 ( second and fourth entry of an angleBonds contribution ). The contribution to the dynamical matrix is calculated for an energy given by anglestrenght*(angle-angle_0)**8
astar=2*math.pi/3.95 cstar=2*math.pi/12.06556 eps=0.0011 Q=Numeric.array( [ [astar*0.5*i+eps,-astar*0.5*i+eps,0] for i in range(1,2)] ) debyerange=[ -12, -12, -6] debye=astar/4 SMcellrange=[2,2,2] cellrange= [-1,-1,-1] Kcellrange=[-6,-6,-12] sigmacellrange=[-4,-4,-2] sigmacharge=0.5 IS_IT_SCREENED=0 USE_SCREENING_FUNCTION =1 ScreenKrange = [6,6,12] selectors=[(1.0,1.0,0.0), (1.0,-1.0,0.0), (1.0,0.0,1.0), (-1.0,1.0,0.0)]
Note the ![]() range that is specified by the variable ScreenKrange.
The contribution from Coulomb interaction without screening function is still
active (note IS_IT_SCREENED=0) but gives zero contributions
as Kcellrange and sigmacellrange are set to zero.
one could still calculate both contribution if the inputted screening function
is
range that is specified by the variable ScreenKrange.
The contribution from Coulomb interaction without screening function is still
active (note IS_IT_SCREENED=0) but gives zero contributions
as Kcellrange and sigmacellrange are set to zero.
one could still calculate both contribution if the inputted screening function
is
The screening function must be set in the parinputedit with the following format :
def SCREENING_FUNCTION(self, Q): moduli2 = Numeric.sum(Numeric.transpose(Q*Q)) astar=2*math.pi/3.95 debye=astar/4 debye=debye*debye # res=(1.0/(1.0+debye/moduli2) )-1 # debye=0.0 res=(1.0/(1.0+debye/moduli2) ) return resThats just an example. You can write whatever function you whish provided that you return either an array of scalars or an array of
The dynamical structure factor could be obtained from the inelastic scattering
of the X-ray under the following conditions [9]:
i) the scattering process is dominated by the Thompson term and there is
no electronic excitation in the energy transfers region,
ii) adiabatic approximation is valid, so that the center of mass of the
electronic clouds could be identified with the nuclear one.
The first assumption is simultaneously satisfied with X-ray with energy
in the
![]() energy range used for IXS experiments,
when we look at energy transfers
energy range used for IXS experiments,
when we look at energy transfers ![]() (
(![]() ).
The second one is a very general assumption in condensed matter theory.
Under these assumptions we can write the inelastic cross section
for one-phonon process as follows. First, we decompose the moment as
).
The second one is a very general assumption in condensed matter theory.
Under these assumptions we can write the inelastic cross section
for one-phonon process as follows. First, we decompose the moment as
![]() (in the reciprocal
lattice vector
(in the reciprocal
lattice vector ![]() plus the phonon pseudo-momentum
plus the phonon pseudo-momentum ![]() ;
hence we obtain for a given angle
;
hence we obtain for a given angle ![]() and frequency
and frequency
![]() :
:
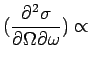 |
|||
| (17) |
where ![]() is the mass for the atom
is the mass for the atom ![]() in the unit cell,
in the unit cell,
![]() is the Debye-Waller factor (which is not calculated in the
present version of OpenPhonon), and
is the Debye-Waller factor (which is not calculated in the
present version of OpenPhonon), and ![]() is the Bose
factor for the mode
is the Bose
factor for the mode ![]() .
The phonon polarization term is
.
The phonon polarization term is
![]() of Eq. 14.
of Eq. 14.
Then the the inelastic cross section can be written as:
|
|||
| (18) |
This corresponds to the neutron scattering cross section for 1-phonon
scattering provided one replaces the nuclear average scattering length
![]() with the atomic form factor
with the atomic form factor
![]() for the atom
for the atom ![]() in the unit cell, and the polarisation factor of the
Thompson scattering
in the unit cell, and the polarisation factor of the
Thompson scattering
![]() .
The latest term is fixed
.
The latest term is fixed ![]() in this version, because most of the
experiments considered where in the scattering confguration with
in this version, because most of the
experiments considered where in the scattering confguration with
![]() [4] or
[4] or
![]() (low
(low ![]() ).
Moreover for high energy resolution inelastic X-ray scattering
we have
).
Moreover for high energy resolution inelastic X-ray scattering
we have
![]() . For a more detailed description
of the IXS cross section, see also the review of E. Burkel [10]
and references therein.
. For a more detailed description
of the IXS cross section, see also the review of E. Burkel [10]
and references therein.
In OpenPhonon the calculation is made calling the program scattering_shifted.py in the following way :
python scattering_shifted.py storeall paramfilewhere paramfile is the name of a file like this :
BrillShift_List=[(6,0,0)]
scatt_dictio_f0={ 'Cu_G0':'Cu1+', 'O_G0':'O','O_G1':'0',\
'Nd_G0':'Nd','Nd_G1':'Nd' }
scatt_dictio_f12={ 'Cu_G0':'Cu', 'O_G0':'O','O_G1':'0',\
'Nd_G0':'Nd','Nd_G1':'Nd' }
Lambda=0.783867
Temp = 15.0
A sample input file is delivered in the distribution archive under the name
scatt_shinput.
The intensities are calculated over the set of points defined by the variable
The atomic scattering factors are read using the Dabax database. The ![]() factors
are read from WaasKirf compilation and f1,f2 from Windt datafile.
factors
are read from WaasKirf compilation and f1,f2 from Windt datafile.
To specify the scattering factors of the atoms one has to specify the wavelength
of X-rays and the two dictionaries, one for ![]() and the other for (f1,f2).
The dictionary keywords are the equivalence class names and their
values are Dabax record names like "Cu" for neutral copper or "Cu1+" for
copper ion. These names must exist in the database otherwise an error
occurs.
Finally the temperature ( in Kelvin) has to be entered to properly
take into account the Bose-Einstein statistics.
and the other for (f1,f2).
The dictionary keywords are the equivalence class names and their
values are Dabax record names like "Cu" for neutral copper or "Cu1+" for
copper ion. These names must exist in the database otherwise an error
occurs.
Finally the temperature ( in Kelvin) has to be entered to properly
take into account the Bose-Einstein statistics.
The results are stored in the file scatt_results
in the form:
Qx,Qy,Qz,frequency(1),intensity_stoke(1),intensity_antistoke(1),..,
frequency(j),intensity_stoke(j),intensity_antistoke(j),..,
frequency(N),intensity_stoke(N),intensity_antistoke(N)
where j indicates the mode in increasing
![]() frequency order.
frequency order.
PDOS is constructed as a N points histogram,
whose frequency coordinates range from zero up to the maximum
eigen-frequency of the system. The histogram bins are filled calculating
the eigen-frequencies over the Brillouin zone
on a
![]() grid.
Symmetries are used to reduce the number of needed calculations.
To get the partial DOS for a given site group,
the contributions are weighted when added on a particular bin.
The weight is calculated as the modulus square
of the eigenvector projection over the considered
degrees of freedom.
grid.
Symmetries are used to reduce the number of needed calculations.
To get the partial DOS for a given site group,
the contributions are weighted when added on a particular bin.
The weight is calculated as the modulus square
of the eigenvector projection over the considered
degrees of freedom.
The calculation of the frequencies in all the point of the grid is performed running the program generapuntiDeb.py:
openphonon generapuntiDeb.py parinput cella generapuntiinp
where the files parinput and cella are the same of section 4.1, and the file generapuntiinp is a file as generapuntiDebinput in the examples directory, with the following structure:
IS_IT_SCREENED=1
Kcellrange=[8,8,2]
sigmacellrange=[8,8,2]
cellrange= [-1,-1,-1]
debyerange=[8,8,2]
SMcellrange=[2,2,2]
astar=2*math.pi/3.95
kdebye=astar/4
eps=0.001
Nsampling=21
Qall=Numeric.array( [ Numeric.array([0.00001,0,0]) + qx*cella.Brillvectors[0]*0.5/(Nsampling-1)
+ qy*cella.Brillvectors[1]*0.5/(Nsampling-1) + qz*cella.Brillvectors[2]*0.5/(Nsampling-1)
for qx in range(0,Nsampling)
for qy in range(0 , Nsampling)
for qz in range(0,Nsampling)
] )
thetasym=45
bdx=math.cos(thetasym*math.pi/180.)
bdy=math.sin(thetasym*math.pi/180.)
basedomain=Numeric.array([ bdx, bdy ,1])
domains=Numeric.array( [
[ bdx, bdy ,1],
.............................
.............................
.............................
where Nsampling correspond to the grid density ![]() .
.
Subsequently, the number of state per bin is calculated by running the program pa_dos.py:
openphonon pa_dos.py storeall Number_of_histograms
where storeall is the one created by generapuntiDeb.py, and Number_of_histograms = N as described above.
Rendering_Radii=[0.5,0.6,0.7 ] colors=[ [0.0,0.0,1.0], [1.0,0.0,0.0], [0.0,0.5,0.0] ] nneig=[1,1,1]
the Rendering_Radii are the visualised sphere radii for the atomic species
defined in <structure_file>. The colors are defined as RGB triplets, ![]() being
the brightest value.
The variable
being
the brightest value.
The variable ![]() specifies how many times the unit cell has to be repeated in
specifies how many times the unit cell has to be repeated in ![]() directions,
directions,
![]() meaning just the central one. For example,
meaning just the central one. For example, ![]() specifies
a range spanning from
specifies
a range spanning from ![]() to
to ![]() for each direction.
The display window can be popped up using the following command :
for each direction.
The display window can be popped up using the following command :
<pymol.com> structureRendering.py <rendering-file-name> \ <structure_file-name>
where pymol.com is the ![]() interpreter.
interpreter.
The example given in the test_OP directory ( binary distribution ) can be runned using the openPhonon wrapper in the following way :
openphonon structureRendering.py render preparationinput
The visualisation of eigenmodes is done via structure rendering.
<pymol.com> eigenRendering.py <rendering-file-name> \
<structure_file-name> <storeall_name>
where storeall is the output file of dispersionDeb.py and rendering-file has the additional information of which mode has to be visualised :
Rendering_Radii=[0.5,0.6,0.7 ] colors=[ [0.0,0.0,1.0], [1.0,0.0,0.0], [0.0,0.5,0.0] ] nneig=[1,1,1] NPscan=48 NPvector=41 fampli= 2.0
where NPscan specifies ![]() ( NPscan starts from 0)
( NPscan starts from 0)
and NPvector is the eigenvector number ( NPvector goes from 0 up
to ![]() is).
is).
python scattering_shifted.py storeall paramfile
one types :
openphonon scattering_shifted.py storeall paramfile
The pymol interpreter is called also through openphonon as :
openphonon eigenRendering.py <rendering-file-name> <structure_file-name>\ <storeall_name>
Installation is done by downloading the distribution archive
(www.esrf.fr/computing/scientific/)
and following the instructions therein.
One needs Python2.X, a C++ compiler, and the NumPy extension package linked with lapack3.0 .
Alternatively one can download the following file containing a self contained tree of binary codes (pre-compiled version, only for linux):
ftp.esrf.fr/pub/scisoft/ESRF_sw/opbin.tgz
This binary distribution should work with Suse or Redhat ![]() .
One downloads it on the root directory of
a Linux machine. It expands as /opt/ESRF_sw.
Binaries and libraries are in /opt/ESRF_sw/bin and /opt/ESRF_sw/lib respectively.
You have to set PATH and LD_LIBRARY_PATH accordingly.
Test files are in /opt/ESRF_sw/test_OP.
Then one should be able to call the openphonon wrapper. Then one
replaces the command python by openphonon.
Pymol is given along with the binary distribution. The pymol home page is
at www.pymol.com.
.
One downloads it on the root directory of
a Linux machine. It expands as /opt/ESRF_sw.
Binaries and libraries are in /opt/ESRF_sw/bin and /opt/ESRF_sw/lib respectively.
You have to set PATH and LD_LIBRARY_PATH accordingly.
Test files are in /opt/ESRF_sw/test_OP.
Then one should be able to call the openphonon wrapper. Then one
replaces the command python by openphonon.
Pymol is given along with the binary distribution. The pymol home page is
at www.pymol.com.
If you are installing from sources be warned that Numpy might need to be linked against lapack3.0. On some machine the numpy provided lapack_lite give problems with Heigenvectors routine that is a kay ingredient of openphonon. It is easy to correct eventual problem by linking against lapack library, one has to modify the numpy setup.py makefile in the following way ( adapt directories and library name to your lapack installation ):
# delete all but the first one in this list if using your own LAPACK/BLAS
sourcelist = [os.path.join('Src', 'lapack_litemodule.c'),
# os.path.join('Src', 'blas_lite.c'),
# os.path.join('Src', 'f2c_lite.c'),
# os.path.join('Src', 'zlapack_lite.c'),
# os.path.join('Src', 'dlapack_lite.c')
]
# set these to use your own BLAS
library_dirs_list = ["/scisoft/ESRF_sw/linux_i386/lib"]
libraries_list = [ "lapack","blas" ]
This document was generated using the LaTeX2HTML translator Version 2002-2-1 (1.70)
Copyright © 1993, 1994, 1995, 1996,
Nikos Drakos,
Computer Based Learning Unit, University of Leeds.
Copyright © 1997, 1998, 1999,
Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The command line arguments were:
latex2html -split 0 manual.tex
The translation was initiated by Alessandro Mirone on 2006-03-21
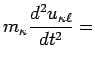
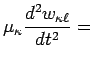
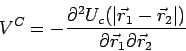
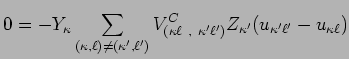
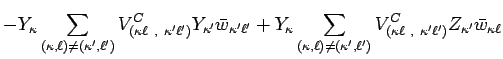
![$\displaystyle -K_x \bar w_{\kappa \ell }
- \sum_{(\kappa,\ell)\neq(\kappa',\ell...
...)}^{S} +
V_{(\kappa \ell ~,~\kappa ' \ell ' )}^{T'}
\Bigl] u_{\kappa ' \ell ' }$](img53.png)
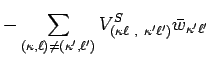
![$\displaystyle \sum_{(\kappa,\ell)\neq(\kappa',\ell')}\Bigl[
V_{(\kappa \ell ~,~...
...l ~,~\kappa ' \ell ' )}^{T'}
\Bigl] ( u_{\kappa \ell } + \bar w_{\kappa \ell })$](img55.png)