- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Structural biology
- Endo-α-mannosidase: an alternative route to N-glycan processing
Endo-α-mannosidase: an alternative route to N-glycan processing
Asparagine-linked (N-linked) glycans direct the folding and targeting, as well as influencing the stability and function of mature glycoproteins. Transfer and biosynthetic modification of a conserved 14-oligosaccharide precursor occurs within the endoplasmic reticulum and features the sequential hydrolysis and transfer of individual glycoside moieties. Aberrant glycosylation, through modification of this process, is strongly associated with cellular dysfunction and disease, including the metastatic progression of cancer and congenital disorders. Furthermore, N-glycans are known to play significant roles in viral proliferation, rendering the various enzymes involved in their maturation as potential therapeutic targets.
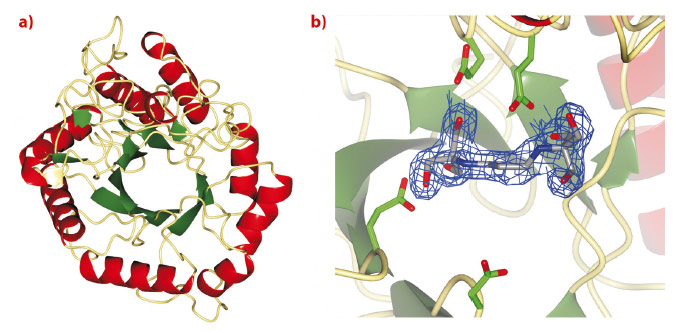
Treatment strategies to regulate diseases of glycoprotein biosynthesis have, to date, focussed largely on the use of inhibitors of the ‘trimming’ glycosidases acting early in the N-glycan processing pathway, such as glucosidase I. The limited efficacy of such compounds to provide effective disease control has arisen, in part, due to the existence of a unique “bypass” enzyme acting within the same pathway, endo-a-mannosidase. Endo-a-mannosidase circumvents the well-known N-glycan processing pathway, specifically hydrolysing an internal bond within the precursor oligosaccharide to liberate a mannose moiety flanked by at least one glucosyl moiety. Using ESRF data, we have recently revealed the first native 3-D structures of a bacterial endo-a-mannosidase enzyme (Figure 11a). Data collected at beamline ID14-1 revealed an initial protein-ligand complex allowing the first view of the enzyme active site (Figure 11b).
 |
|
Fig. 11: a) 3-D structure and b) electron density for BIS-TRIS-Propane bound in the endo-a-mannosidase active site, both determined using data collected at the ESRF. |
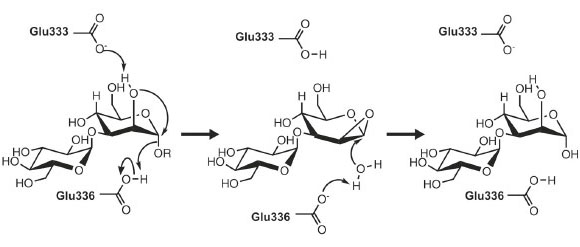
As might be expected given its unique activity, the 3-D structure of endo-a-mannosidase appears relatively novel compared to those within the 120+ currently known glycoside hydrolase families. The active site appears to be located within a long solvent-accessible cleft, consistent with the linear, elongated nature of the N-glycan substrate. The majority of amino acids comprising this region are highly conserved among endo-α-mannosidases from different species. Highly unusual within these structures is the apparent absence of divalent metal ions, or even a site available for metal coordination, a feature typically synonymous with a-mannosidase activity. These observations, together with further structural complexes and kinetic analysis, have led us to propose a novel, substrate-assisted mechanism for catalysis via a 1,2 epoxide intermediate (Figure 12).
 |
|
Fig. 12: Potential endo-α-mannosidase reaction mechanism via an epoxide-like 1,2-anhydro sugar intermediate. |
Given that detailed knowledge of enzymatic mechanisms is of central importance to the rational design of inhibitors and potential drug compounds, future work will focus on the complete mechanistic dissection of endo-α-mannosidase activity, together with further structural analysis of complexes. Inhibitors that block the early processing stages of N-glycan biosynthesis have been shown to provide some promise as novel treatment strategies to a wide range of pathologies including cancer and viral infections. As such, the elucidation of the structure of endo-a-mannosidase provides a valuable rationale for the development of novel compounds, which might form part of combinatorial antiviral therapies together with existing a-glucosidase I/II inhibitors.
Principal publication and authors
A.J. Thompson (a), R.J. Williams (b), Z. Hakki (b), D.S. Alonzi (c), T. Wennekes (d), T.M. Gloster (a), K. Songsrirote (a,e), J.E. Thomas-Oates (a,e), T.M. Wrodnigg (f), J. Spreitz (f), A.E. Stütz (f), T.D. Butters (c), S.J. Williams (b) and G.J. Davies (a), Proc. Natl. Acad. Sci. (USA) 109, 781-786 (2012).
(a) Department of Chemistry, University of York (UK)
(b) School of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne (Australia)
(c) Oxford Glycobiology Institute, Department of Biochemistry, University of Oxford (UK)
(d) Laboratory of Organic Chemistry, Wageningen University (The Netherlands)
(e) Centre of Excellence in Mass Spectrometry, University of York (UK)
(f) Institute of Organic Chemistry, Graz University of Technology (Austria)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.