- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2009
- Dynamics and extreme conditions
- The electronic structure of sulphur studied by X-ray absorption and emission spectroscopy
The electronic structure of sulphur studied by X-ray absorption and emission spectroscopy
Sulphur forms chemical bonds with atoms with very different electronegativity values resulting in large variations of its local electronic structure. This polyvalence makes sulphur an abundant element that plays a key role in various fields such as in catalytic and biological processes and in environmental systems. The chemical state of sulphur has therefore been widely studied by means of XANES (X-ray absorption near edge structure) spectroscopy [1]. XANES is a powerful tool to study the density of unoccupied electronic states with element selectivity. However, the strong influence of the number and type of ligands and the local geometry as well as possible experimental artefacts (e.g. self-absorption in fluorescence detected XANES) may lead to difficulties in the interpretation of the XANES spectra. The chemical dependence is considerably simpler in Ka X-ray emission spectroscopy (XES) than in XANES [2]. In this work we report a comprehensive experimental and theoretical study of the K![]() fluorescence lines in a variety of sulphur compounds with a critical comparison with the corresponding XANES spectra. The measurements were carried out at beamline ID26.
fluorescence lines in a variety of sulphur compounds with a critical comparison with the corresponding XANES spectra. The measurements were carried out at beamline ID26.
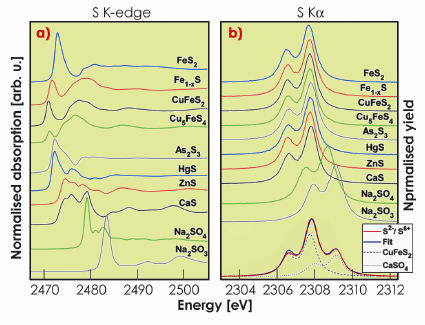
Figure 14a shows the XANES spectra of various sulphur compounds. The profound variations in the spectral shape reflect the strong influence of the local structure and the type of cation on the orbital hybridisation around the sulphur atom. The absorption edge position depends on the oxidation state via charge screening effects and a shift of 12 eV (Figure 15a) is observed between sulphides (S in oxidation state -2) and sulphates (S in oxidation state +6). In sulphides, the sulphur atom is bonded directly to the metal cation and the resulting bonding character of the sulphide has a considerable influence on the position of the S K-edge. The energy position of the S K![]() XES - arising from 2p to 1s transition - reflects the chemical state by a similar screening effect that changes the core level binding energy but the spectral shape is little sensitive to the chemical environment (Figure 14b). The S K
XES - arising from 2p to 1s transition - reflects the chemical state by a similar screening effect that changes the core level binding energy but the spectral shape is little sensitive to the chemical environment (Figure 14b). The S K![]() 1 position shows an energy shift of 1.5 eV with the change in the formal oxidation state (Figure 15a). Even though the spectral changes on an absolute scale are an order of magnitude smaller in XES than in XANES, a very detailed analysis of the energy position is possible because the Ka energy can be measured with high accuracy and the relative peak intensity is barely influenced by self-absorption in the sample. The absence of strong interactions between the 2p core hole and the valence electrons in the K
1 position shows an energy shift of 1.5 eV with the change in the formal oxidation state (Figure 15a). Even though the spectral changes on an absolute scale are an order of magnitude smaller in XES than in XANES, a very detailed analysis of the energy position is possible because the Ka energy can be measured with high accuracy and the relative peak intensity is barely influenced by self-absorption in the sample. The absence of strong interactions between the 2p core hole and the valence electrons in the K![]() XES makes the screening effect the dominant source of the chemical sensitivity. The simple chemical sensitivity provides an opportunity for a quantitative analysis in heterogeneous systems. A sulphide/sulphate mixture with 66.7% of sulphur present in the chemical state S2- is shown in Figure 14b. Fitting of the experimental data with a weighted sum of representative spectra for S2- and S6+ gives a contribution of 66.3% from the S2- spectrum, demonstrating that XES can be used to quantitatively determine the amount of sulphur species in mixed-valence systems.
XES makes the screening effect the dominant source of the chemical sensitivity. The simple chemical sensitivity provides an opportunity for a quantitative analysis in heterogeneous systems. A sulphide/sulphate mixture with 66.7% of sulphur present in the chemical state S2- is shown in Figure 14b. Fitting of the experimental data with a weighted sum of representative spectra for S2- and S6+ gives a contribution of 66.3% from the S2- spectrum, demonstrating that XES can be used to quantitatively determine the amount of sulphur species in mixed-valence systems.
 |
|
Fig. 14: a) S K-edges and b) S K |
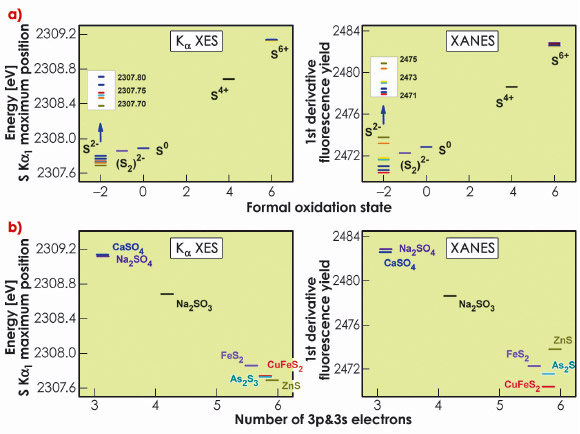
The energy positions of the sulphides (S-2) are spread over a large range. The ordering within the sulphides is different between XES and XANES. We investigated this discrepancy by comparing the experimental results to the charge density as extracted from quantum chemical calculations. Figure 15a shows the energy positions of the K![]() 1 emission line and K absorption edge as a function of the calculated number of 3p and 3s electrons. This approach shows a monotonic relation between the K
1 emission line and K absorption edge as a function of the calculated number of 3p and 3s electrons. This approach shows a monotonic relation between the K![]() 1 line position and the sulphur charge, in particular within the series of sulphides, which is not observed for the XANES edge position.
1 line position and the sulphur charge, in particular within the series of sulphides, which is not observed for the XANES edge position.
 |
|
Fig. 15: S K |
We find that XES can serve as a probe of the local charge density with considerably higher accuracy than XANES. The more complex XANES spectral shape arises from its sensitivity to a multitude of mechanisms. The more selective sensitivity of K![]() XES greatly facilitates interpretation of experimental results. Comparison with calculations shows that the K
XES greatly facilitates interpretation of experimental results. Comparison with calculations shows that the K![]() 1 energy can be correlated to high accuracy with the valence-shell electron population and good agreement is achieved for the Ka lines. Furthermore, the selectivity of XES can be used to quantify the amount of sulphur species in heterogeneous systems and its simple spectral shape renders such a quantitative analysis considerably more robust than a similar approach in XANES.
1 energy can be correlated to high accuracy with the valence-shell electron population and good agreement is achieved for the Ka lines. Furthermore, the selectivity of XES can be used to quantify the amount of sulphur species in heterogeneous systems and its simple spectral shape renders such a quantitative analysis considerably more robust than a similar approach in XANES.
References
[1] E. Solomon et al., Coord. Chem. Rev. 249, 97 (2005).
[2] M. Kavcic et al., Nucl. Instrum. Meth. B 222, 601 (2004).
Principal publication and authors
R. Alonso Mori (a,b), E. Paris (b), G. Giuli (b), S.G. Eeckhout (a), M. Kavčič (c), M. Žitnik (c), K. Bučar (c), L.G.M. Pettersson (d) and P. Glatzel (a), Anal. Chem. 81, 6516 (2009).
(a) ESRF
(b) Dipartamento di Scienze della Terra, Camerino University (Italy)
(c) J. Stephan Institute (Slovenia)
(d) FYSIKUM, Stockholm University (Sweden)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.