- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2006
- X-ray Absorption and Magnetic Scattering
- Negative thermal expansion and local dynamics in Cu2O and Ag2O
Negative thermal expansion and local dynamics in Cu2O and Ag2O
While most crystals expand when temperature is increased, some framework structures, composed by networks of corner-sharing polyhedral structural units, exhibit negative thermal expansion (NTE) extending over temperature intervals of several hundreds of Kelvin; the most popular example is ZrW2O8, where NTE extends up to 1050 K. The macroscopic thermal expansion is generally considered the result of competition between a positive contribution due to bond stretching and a negative contribution of geometrical origin, like bond bending, which in framework structures is often connected to low-frequency rigid unit modes (RUMs) which cause rigid rotations of the basic polyhedral units. While it is generally accepted that thermal vibrations perpendicular to inter-atomic bonds play a key role in NTE, a satisfactory quantitative understanding of the local mechanisms is still lacking [1].
New information on the origin of NTE can be gained by combining Bragg diffraction and EXAFS, and exploiting the different sensitivity of the two techniques to atomic vibrations and thermal expansion. Bragg diffraction measures the distances between average atomic positions and the atomic thermal factors of individual atoms (MSDs), EXAFS is sensitive to the average bond distances and measures the variance of the bond-length distribution (parallel MSRD). The thermal expansions measured by EXAFS and diffraction are different, and their comparison allows one to evaluate the MSRD perpendicular to the bond length [2].
 |
|
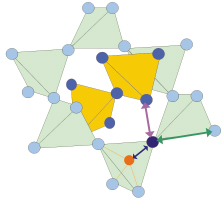
Fig. 103: The cuprite structure is made by two interpenetrating networks of corner sharing M4O tetrahedra (M=Cu, Ag). |
Copper(I) and Silver(I) oxides, Cu2O and Ag2O, are particularly well suited to study the NTE mechanism. Both compounds share the same simple cuprite structure, made by two interpenetrating lattices, one fcc of metal atoms, one bcc of oxygen atoms, which can alternatively be considered as formed by two interpenetrating networks of corner-sharing M4O tetrahedra (M=Cu,Ag); each M atom is linearly coordinated to two O atoms, and each O atom is tetrahedrally coordinated to four M atoms (Figure 103). Both compounds are characterised by NTE extending over large temperature intervals, although with different extents and temperature dependencies (Figure 104).
 |
|
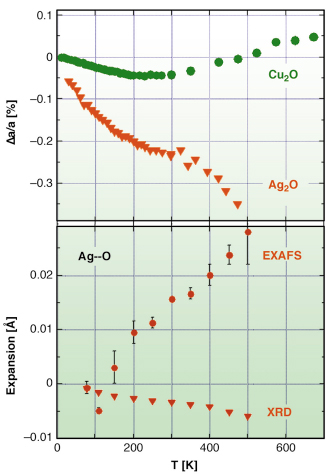
Fig. 104: Relative lattice expansions of Cu2O and Ag2O measured by Bragg diffraction (top panel) and comparison between Ag-O distance expansion measured by Bragg diffraction and EXAFS (triangles and circles, respectively, in bottom panel). |
Cu2O and Ag2O have been studied at the ESRF by diffraction (beamlines BM16 and ID31) and EXAFS (BM08 and BM29). According to diffraction, thermal expansion in Cu2O is negative below 240 K, and becomes positive above 300 K, while in Ag2O it is negative up to at least 470 K, and much stronger than in Cu2O. The thermal factors of M atoms are anisotropic, characterised by a large vibrational amplitude perpendicular to the O-M-O linear link.
EXAFS shows that the nearest neighbour M-O distance (dark blue arrow in Figure 103, and bottom panel in Figure 104) undergoes positive expansion in both compounds. Moreover, the stronger the lattice negative expansion (Ag2O versus Cu2O), the larger the positive M-O expansion and the perpendicular MSRD. Further information was gained by comparing the EXAFS MSRDs with the diffraction MSDs. The correlation of atomic motion in the parallel direction is near its maximum possible value, while a weaker correlation affects the perpendicular motion, indicating that the M-O bond is much stiffer towards stretching than towards bending, and that the perpendicular to parallel anisotropy of relative thermal motion (EXAFS) is stronger than the anisotropy of the absolute motion of Cu and Ag atoms (diffraction).
A refined second-shell analysis led us to distinguish the different behaviour of the two kinds of M--M pairs, having the same crystallographic bond-lengths but belonging to the same or to different networks of M4O tetrahedra (green and pink arrows in Figure 103, respectively): negative and positive expansion is observed for the intra-network and inter-network pairs, respectively, the former undergoing a weaker parallel relative motion than the latter.
These results reveal a complex local behaviour in a relatively simple structure, the correlation of vibrational motion playing a key role. The intense relative motion of second-shell M--M atomic pairs indicates a strong deformation of the M4O tetrahedra, ruling out the possibility of explaining NTE in cuprite structures by a simple RUM model. The stiffness of the M-O bonds suggests that an alternative model for NTE in cuprites could be based on the more flexible idea of rigid M-O rods.
References
1] G.D. Barrera, J.A. Bruno, T.H.K. Barron and N.L. Allan, J. Phys.: Condens. Matter 17, R217 (2005).
[2] P. Fornasini, S. a Beccara, G. Dalba, R. Grisenti, A. Sanson, M. Vaccari and F. Rocca, Phys. Rev B 70, 1 (2004).
Principal Publications and Authors
A. Sanson (a), F. Rocca (a), G. Dalba (b), P. Fornasini (b), R. Grisenti (b), M. Dapiaggi (c) and G. Artioli (c), Phys. Rev B 73, 214305 (2006).
(a) IFN-CNR, Trento (Italy)
(b) INFM & Dip.Fisica, Univ. Trento (Italy)
(c) Dip. Scienze della Terra, Univ. Milano (Italy)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.