- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2006
- X-ray Absorption and Magnetic Scattering
- Synchronising subsecond infrared and X-ray absorption spectroscopies to see inside a reactive process
Synchronising subsecond infrared and X-ray absorption spectroscopies to see inside a reactive process
Catalytic reactions mediated by heterogeneous catalysts are the hub of many important processes; the abatement of toxic emissions from car exhausts is a prime example. To this day, catalysts based upon a Rh component supported on an Al2O3 dispersant can be found in practically any petrol driven car. Rh is present as it has a unique capacity for so called “de NOx” chemistry, wherein toxic species such as NO and N2O are removed from the exhaust and replaced by N2.
The details of how this conversion is mediated by Rh are, however, far from complete. Part of the reason for this is that such systems have never been studied in a manner that addresses their structural, functional, and reactive character at the same time, and with sufficient speed to see inside rapid processes. To this end a new experiment has been developed and implemented on ID24. This fuses a reactivity probe (mass spectrometry), a probe of surface functionality (diffuse reflectance infrared spectroscopy (DRIFTS)), and a time resolving probe of local structure (energy-dispersive extended X-ray absorption fine structure), to produce a methodology that can probe a reactive system in a more holistic manner.
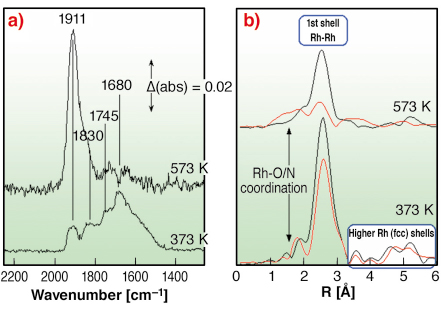
Figure 97 shows data typical of the DRIFTS (a) and dispersive EXAFS (b) components of the experiment. DRIFTS indexes the presence of a range of molecular NO species adsorbed on the surface of the catalyst, e.g.: linear (NO+), 1911 cm-1; bent (NO-), 1745 cm-1; geminal (Rh(NO)2), 1830 and 1745 cm-1; and NO associated with metallic Rh surfaces (1680 cm-1).
 |
|
Fig. 97: a) DRIFTS spectra, acquired in 64 milliseconds, recorded after 50 seconds exposure of a 5wt%Rh/Al2O3 catalyst to 5%NO/He at the temperatures indicated. b) Fourier Transform representation of dispersive EXAFS data, acquired in 62 milliseconds, before reaction (black) and after (red) NO exposure. |
Dispersive EXAFS reveals how the local Rh structure changes in response to NO adsorption. At 373 K the Rh remains face centred (fcc), as indicated by the persistence of higher Rh shells that follow the expected radial progression for this structure. The average particle size (from the first shell Rh-Rh intensity) appears diminished as a result of molecular NO adsorption. At 573 K the interaction with NO changes the phase of the Rh. At this temperature NO dissociation predominates and the formation of an oxidic form of Rh results.
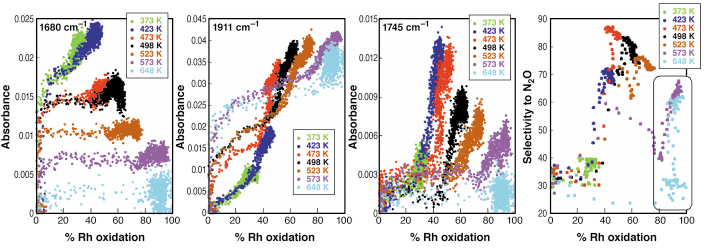
In between these points, a variety of structural-reactive chemistry has occurred. Some specific aspects of this “hidden” detail are shown in Figure 98. In this figure, for several temperatures, and for three of the observed NO species, the DRIFTS intensity (in absorbance units) is plotted as a function of Rh oxidation (from EXAFS). Additionally, the far right panel shows how the net selectivity of the interaction of NO varies with Rh oxidation.
 |
|
Fig. 98: Correlation plots relating Rh oxidation to surface nitrosyls and selectivity. 100% Rh oxidation is assumed as the attainment of an Rh2O3 stoichiometry. |
A clear correlation emerges between the bent nitrosyl species (1745 cm-1) and the selectivity, suggesting that it is this form of NO that plays a predominant role in the production of N2O. Moreover, this species is only supported by the Rh in significant amounts over a relatively narrow range of Rh oxidation. Other forms of NO, associable with either very reduced (1680 cm-1) or very oxidised (1911 cm-1) Rh sites appear not, by and large, to have any great relation to the most active route for the production of N2O from NO.
That said, the experiment also shows that a second route to N2O can exist, but only at high T and high levels of Rh oxidation. The increase in selectivity for N2O (circled in black) observed in these cases links this second mechanism to the persistence of the linear nitrosyl (1911 cm-1) on an oxidic Rh phase. Addition of an NO(g) molecule, to this nitrosyl species can form N2O directly, whilst simultaneously adding an O atom to, and removing a defect site from, the growing oxidic phase.
In summary, by combining time resolved EXAFS with infrared spectroscopy and mass spectrometry we have been able to see inside a fundamental gas-solid interaction and begun to reveal the parameterisation that determines how a catalyst mediates the conversion of a given substrate molecule to one or other possible end result.
This research was funded by the EPSRC UK (Grant Number GR/60744/01) and carried out under a Long Term Proposal award from the ESRF.
Principal Publications and Authors
M.A. Newton (a), A.J. Dent (b), S.G. Fiddy (c), B. Jyoti (d), J Evans (d), Catal. Today, doi: 10.1016/j.cattod. 2006.09.034., Phys. Chem. Chem. Phys. 9, 246-249 (2007); J. Mater Chem., (2006) in press.
(a) ESRF
(b) Diamond Light Source (UK)
(c) SRS Daresbury laboratory (UK)
(d) School of Chemistry, University of Southampton, and Diamond Light Source (UK)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.