- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2004
- Structural Biology
- Loading the Calcium Pump
Loading the Calcium Pump
Cation pumps maintain the electrochemical gradients of cations across biomembranes. These membrane proteins are also known as P-type ATPases due to their use of a phosphoenzyme intermediate in a functional cycle driven by ATP hydrolysis[1]. Prominent examples of cation pumps in higher organisms include the Na+,K+-ATPases, which maintain the membrane potential in animals and the ability to perform secondary transport, the H+,K+-ATPases which are the acid pump of the stomach, and the Ca2+-ATPases, which maintain calcium levels as required for the use of this cation in signalling. We are studying rabbit SERCA1a, a 110 kDa Ca2+-ATPase isolated from the membrane of sarco(endo)plasmic reticulum from fast-twitch muscle. This protein pumps Ca2+ from the cytoplasm to the lumen, thereby terminating the activity of myosin-actin filaments. It represents an attractive model system of the P-type ATPases. Our aim is to obtain crystal structures of its defined functional states and thus to address the general question on how Nature has devised a cation pump running on ATP.
The functional cycle of SERCA, and the P-type ATPases in general, is typically depicted by E1 and E2 states. A Ca2E1 state is adopted upon binding two cytoplasmic Ca2+ ions [2]. When further supplied with ATP, ![]() -phosphoryl transfer to the conserved Asp351 residue takes place and the Ca2E1~P state is reached representing the high-energy intermediate state of the cycle. Formation of this state is coupled with the occlusion of the two bound Ca2+ ions, thereby preventing an unprofitable backflow to the cytoplasm once energy is being spent. This coupling mechanism is probably general in the P-type ATPases. From the Ca2E1~P state the cycle proceeds through downhill transitions associated with the release of the two Ca2+ ions on the other side of the membrane in the E2P state, binding of counter-cation (protons for Ca-ATPase) and an auto-catalysed dephosphorylation leading to the E2 state.
-phosphoryl transfer to the conserved Asp351 residue takes place and the Ca2E1~P state is reached representing the high-energy intermediate state of the cycle. Formation of this state is coupled with the occlusion of the two bound Ca2+ ions, thereby preventing an unprofitable backflow to the cytoplasm once energy is being spent. This coupling mechanism is probably general in the P-type ATPases. From the Ca2E1~P state the cycle proceeds through downhill transitions associated with the release of the two Ca2+ ions on the other side of the membrane in the E2P state, binding of counter-cation (protons for Ca-ATPase) and an auto-catalysed dephosphorylation leading to the E2 state.
We have studied the transition to the Ca2E1~P state by determining the crystal structures of calcium-bound complexes with i) the non-hydrolysable ATP analogue AMPPCP, and ii) ADP:AlF4- which mimics the ![]() -phosphoryl transfer reaction. Data extending to 2.6 Å resolution on the Ca2E1-AMPPCP form was obtained at beamline ID29 allowing proper density modification of experimental phases (derived from a lead-derivative crystal), and refinement of the structure. Our biochemical data further show that the ADP:AlF4- complex mimics the true Ca2E1~P state very closely, as indicated by the efficient occlusion of bound C2+. Based on the crystal structures we are therefore in a position to study the general mechanism of phosphoryl transfer and its coupling to cation occlusion in the P-type ATPases.
-phosphoryl transfer reaction. Data extending to 2.6 Å resolution on the Ca2E1-AMPPCP form was obtained at beamline ID29 allowing proper density modification of experimental phases (derived from a lead-derivative crystal), and refinement of the structure. Our biochemical data further show that the ADP:AlF4- complex mimics the true Ca2E1~P state very closely, as indicated by the efficient occlusion of bound C2+. Based on the crystal structures we are therefore in a position to study the general mechanism of phosphoryl transfer and its coupling to cation occlusion in the P-type ATPases.
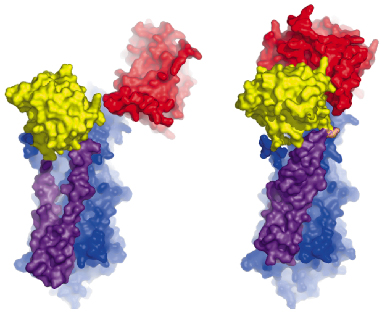 |
Fig. 67: Comparison of the Ca2E1 state (left [2]) and the Ca2E1~P state (right). The A- and N-domains are shown in red and yellow, respectively, and the P-domain as well as the M3-M10 segment in blue. The mobile M1-M2 segment is shown in purple. The large conformational changes result from ATP binding and phosphoryl transfer, and close the cytoplasmic entry to the Ca2+ binding sites. |
The overall structures of the AMPPCP and ADP:AlF4- complexes are similar showing a compact arrangement of the three cytoplasmic domains (A, P and N) relative to the 10 transmembrane segments (M1 through M10) as seen in Figure 67. A comparison with the nucleotide-free Ca2E1 state reveals that large domain movements are induced upon ATP binding leading to a compact arrangement. A closer look at the nucleotide binding pocket in the AMPPCP complex reveals the rationale of phosphoryl transfer as the result of an octahedral coordination to a Mg2+ ion involving the ![]() -phosphate group and the Asp351 side chain as ligands. This arrangement places the side chain of Asp351 in a position for in-line attack (Figure 68). The conformational changes associated with the assembly of the phosphoryl transfer site are transmitted via a rotation of the A-domain to translation of the M1-M2 segments relative to M3 through M10. In this way a cytoplasmic entry to the two Ca2+ binding sites in the membrane is sealed off and occluded by residues of M1 and M2 (Figure 67). Our structure thus explains the coupling mechanism of P-type ATPases.
-phosphate group and the Asp351 side chain as ligands. This arrangement places the side chain of Asp351 in a position for in-line attack (Figure 68). The conformational changes associated with the assembly of the phosphoryl transfer site are transmitted via a rotation of the A-domain to translation of the M1-M2 segments relative to M3 through M10. In this way a cytoplasmic entry to the two Ca2+ binding sites in the membrane is sealed off and occluded by residues of M1 and M2 (Figure 67). Our structure thus explains the coupling mechanism of P-type ATPases.
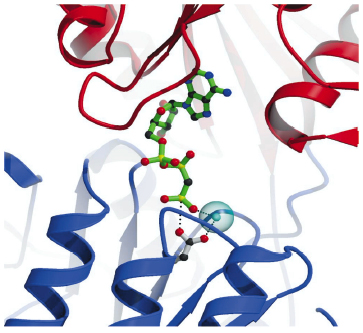 |
Fig. 68: Details of the nucleotide binding pocket in the Ca2E1-AMPPCP complex showing that ATP binding (represented by AMPPCP in green sticks) joins the cytoplasmic N and P-domains (red and blue ribbons, respectively). The |
References
[1] J.V. Møller, B. Juul, M. le Maire, Biochim. Biophys. Acta 1286, 1-51 (1996).
[2] C. Toyoshima, M. Nakasako, H. Nomura, H. Ogawa, Nature 405, 647-655 (2000).
Principal Publication and Authors
T.L. Sørensen, J.V. Møller, P. Nissen, Science 304, 1672-1675 (2004).
University of Aarhus (Denmark)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.