- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2004
- Materials Science
- Heterogeneous Non-classical Cu(I)- and Ag(I)-carbonyls Stabilised in ZSM-5 Zeolite: Thermodynamic and Spectroscopic Features
Heterogeneous Non-classical Cu(I)- and Ag(I)-carbonyls Stabilised in ZSM-5 Zeolite: Thermodynamic and Spectroscopic Features
The electrostatic, ![]() - and -
- and -![]() contributions in the formation of carbonyl bonding for the "non classical" Cu(I)- and Ag(I)- carbonyls hosted inside zeolitic nanocavities have been separated using a combination of thermodynamic and spectroscopic information. Adsorption of CO, at RT, on well-defined Cu(I) and Ag(I)-ZSM-5 systems was studied by the joint use of adsorption microcalorimetry and IR, EXAFS and XANES spectroscopies performed on the beamlines BM8 (GILDA) and BM29, respectively. The formation in the zeolite pores of heterogeneous, [Cu(CO)2]+ and [Ag(CO)]+ complexes, with a stoichiometry that agrees with the homogeneous "non classical" carbonyls, has been observed at pCO = 40 Torr. A C-O stretching frequency higher than that of the free molecule (2143 cm-1) has been observed:
contributions in the formation of carbonyl bonding for the "non classical" Cu(I)- and Ag(I)- carbonyls hosted inside zeolitic nanocavities have been separated using a combination of thermodynamic and spectroscopic information. Adsorption of CO, at RT, on well-defined Cu(I) and Ag(I)-ZSM-5 systems was studied by the joint use of adsorption microcalorimetry and IR, EXAFS and XANES spectroscopies performed on the beamlines BM8 (GILDA) and BM29, respectively. The formation in the zeolite pores of heterogeneous, [Cu(CO)2]+ and [Ag(CO)]+ complexes, with a stoichiometry that agrees with the homogeneous "non classical" carbonyls, has been observed at pCO = 40 Torr. A C-O stretching frequency higher than that of the free molecule (2143 cm-1) has been observed: ![]() CO = 2192 cm-1 for [Ag(CO)]+, while the [Cu(CO)2]+ complexes (Figure 26a) result in a doublet at 2151 and 2178 cm-1 [1-3]. The number of CO molecules adsorbed per Cu(I) cations, obtained by volumetric measurements, agree fairly well with the local stoichiometry emerging from the EXAFS data analysis. As an example, Figure 26b reports the results of the EXAFS data analysis, performed with GNXAS software. Interaction with CO at RT causes a partial solvation of Cu(I) cations from the zeolite walls: the Cu-OF distance (OF = framework oxygen) is elongated by +0.11 ± 0.03 Å. The scattering due to the carbonyl ligands results in a coordination number for the CO molecules of 1.8 ± 0.3, in agreement with the [Cu(CO)2]+ stoichiometry suggested by IR spectroscopy and adsorption microcalorimetry. The Cu-C distance obtained for the [Cu(CO)2]+ complex is 1.88 ± 0.02 Å, while the C-O distance (1.12 ± 0.03 Å) is in good agreement with the gas-phase value (1.128 Å), and the Cu-C-O bond angle is linear within the error bars (170° ± 10°), in agreement with indirect IR evidences.
CO = 2192 cm-1 for [Ag(CO)]+, while the [Cu(CO)2]+ complexes (Figure 26a) result in a doublet at 2151 and 2178 cm-1 [1-3]. The number of CO molecules adsorbed per Cu(I) cations, obtained by volumetric measurements, agree fairly well with the local stoichiometry emerging from the EXAFS data analysis. As an example, Figure 26b reports the results of the EXAFS data analysis, performed with GNXAS software. Interaction with CO at RT causes a partial solvation of Cu(I) cations from the zeolite walls: the Cu-OF distance (OF = framework oxygen) is elongated by +0.11 ± 0.03 Å. The scattering due to the carbonyl ligands results in a coordination number for the CO molecules of 1.8 ± 0.3, in agreement with the [Cu(CO)2]+ stoichiometry suggested by IR spectroscopy and adsorption microcalorimetry. The Cu-C distance obtained for the [Cu(CO)2]+ complex is 1.88 ± 0.02 Å, while the C-O distance (1.12 ± 0.03 Å) is in good agreement with the gas-phase value (1.128 Å), and the Cu-C-O bond angle is linear within the error bars (170° ± 10°), in agreement with indirect IR evidences.
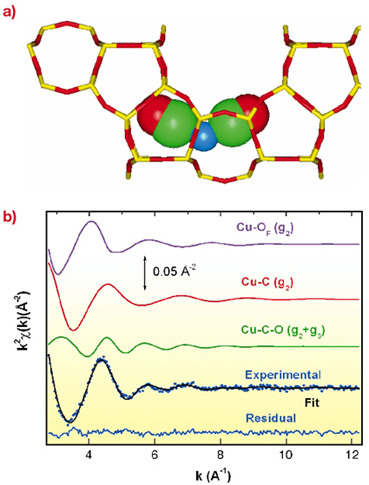 |
Fig. 26: (a) Pictorial representation of a [Cu(CO)2]+ complex (ball representation) hosted inside the ZSM-5 framework (stick representation: O red; Si or Al yellow). (b) The Cu-K edge EXAFS spectrum collected on Cu(I)-ZSM-5 zeolite in interaction with CO at RT (pCO = 40 Torr: blue dotted curve) has been successfully simulated (black full line) as the superimposition of both framework oxygens (magenta line) and two adsorbed CO molecules (red and green curves). |
The zero-coverage enthalpies of CO adsorbed on Cu(I)- and Ag(I)-sites (-![]() adsH ~120 and ~100 kJ/mol, respectively) are much larger than the -
adsH ~120 and ~100 kJ/mol, respectively) are much larger than the -![]() adsH values measured for the Na+ (~35 kJ/mol) and K+ (~28 kJ/mol) sites hosted in the same zeolite pores in spite of the closeness of the charge/radius ratios of the two sets of metal cations [Na+ and Cu(I); K+ and Ag(I)]. The high -
adsH values measured for the Na+ (~35 kJ/mol) and K+ (~28 kJ/mol) sites hosted in the same zeolite pores in spite of the closeness of the charge/radius ratios of the two sets of metal cations [Na+ and Cu(I); K+ and Ag(I)]. The high -![]() adsH values and the irreversible nature of (a fraction of) the d-block metal carbonyls implies the onset of a
adsH values and the irreversible nature of (a fraction of) the d-block metal carbonyls implies the onset of a ![]() -back donation reinforcing the carbonyl bond with respect to a pure s-coordination.
-back donation reinforcing the carbonyl bond with respect to a pure s-coordination.
Insertion of ammonia ligands results in the displacement of one CO molecule and in the formation of intrazeolitic [Cu(NH3)n(CO)]+ complexes characterised by ![]() CO = 2100 cm-1, a value significantly lower than in the gas. This datum indicates that, in such [Cu(NH3)n(CO)]+ complexes, the
CO = 2100 cm-1, a value significantly lower than in the gas. This datum indicates that, in such [Cu(NH3)n(CO)]+ complexes, the ![]() -back donation dominates both in the electrostatic and s-donation components. The charge releasing character of the amino ligand certainly plays a role in quenching the electrostatic component of the Cu(I)
-back donation dominates both in the electrostatic and s-donation components. The charge releasing character of the amino ligand certainly plays a role in quenching the electrostatic component of the Cu(I)![]() CO interaction[1].
CO interaction[1].
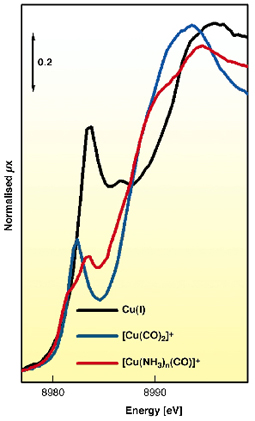 |
Fig. 27: Normalised XANES spectra of Cu(I)-ZSM-5 in vacuo (black line), after interaction with CO on samples activated (blue line) and previously in contact with NH3 (red line). |
Figure 27 reports the important modification of both electronic and geometric configurations of Cu(I) centres upon formation of [Cu(CO)2]+ (blue curve) and [Cu(NH3)n(CO)]+ (red curve) complexes hosted in ZSM-5 zeolite (black curve: zeolite in vacuo). Significant red shift and shape variations of both edge and pre-edge features are observed. XANES data confirm the strong charge releasing character of the amino ligand and explains the low n-CO observed in the IR experiment.
References
[1] A. Zecchina, S. Bordiga, G. Turnes Palomino, D. Scarano, C. Lamberti and M. Salvalaggio, J. Phys. Chem. B, 103, 3833-3844 (1999).
[2] V. Bolis, S. Maggiorini, L. Meda, F. D'Acapito, G. Turnes Palomino, S. Bordiga and C. Lamberti, J. Chem. Phys. 113, 9248-9261 (2000).
[3] C. Lamberti, G. Turnes Palomino, S. Bordiga, G. Berlier, F. D'Acapito and A. Zecchina, Angew. Chem. Int. Ed., 39, 2138-2141 (2000).
Principal Publication and Authors
V. Bolis (a,b), S. Bordiga (b,c), A. Zecchina (b), C. Prestipino (b,c), L. Capello (d), F. D'Acapito (d), C. Lamberti (b,c), J. Phys. Chem. B, 108, 9970-9983 (2004).
(a) University of Piemonte Orientale, and INSTM (Italy)
(b) NIS centre of excellence University of Turin, and INSTM (Italy)
(c) INFM UdR of Turin University (Italy)
(d) ESRF (France)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.