- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2003
- Macromolecular Crystallography
- The Crystal Structure of CDP-ME Kinase: a Target for the Development of Novel Antimicrobial Drugs
The Crystal Structure of CDP-ME Kinase: a Target for the Development of Novel Antimicrobial Drugs
The Crystal Structure of CDP-ME Kinase: a Target for the Development of Novel Antimicrobial Drugs
Due to increasing levels of antibiotic drug resistance there is an urgent need to develop new drugs for microbial infections. Structural biology has much to offer towards this goal by contributing to structure-based design or discovery of potent enzyme inhibitors. The key to the development of novel antibiotics however, is the choice of the enzyme target to be inhibited. Ideally such an enzyme must be essential to pathogens and absent from humans. Recently, a complete metabolic pathway comprising seven enzymes that fulfill this criterion has been delineated [1]. This pathway, termed the 1-deoxy-D-xylulose-5-phosphate (DOXP) pathway, generates isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), the universal precursors of isoprenoids. These are compounds that contribute to key cellular functions including respiration, hormone-based signaling, apoptosis, meiosis, protein cleavage and degradation. They are also components of cell membranes. Organisms that utilise the DOXP pathway include the causal agents for many serious diseases including leprosy, malaria, various gastrointestinal and sexually transmitted infections, trachoma, tuberculosis and certain types of pneumonia. Crucially, mammals utilise a different pathway containing completely different enzymes to make IPP and DMAPP and this makes enzymes in the DOXP pathway very attractive targets for novel antimicrobial drugs.
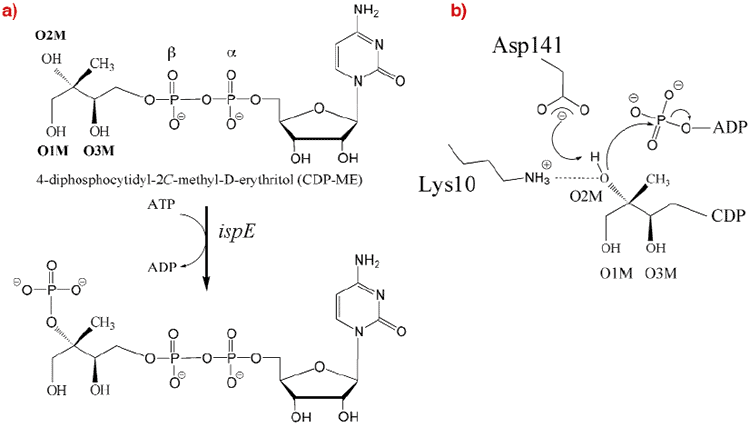
We have thus determined the crystal structure of 4-Diphosphocytidyl-2C-methyl-D-erythritol (CDP-ME) kinase that catalyses the single ATP-dependent phosphorylation stage affording 4-diphosphocytidyl-2C-methyl-D-erythritol-2-phosphate (Figure 28), a reaction at the core of the DOXP pathway.
 |
|
Fig. 28: (a) The reaction catalysed by CDP-ME kinase. (b) The proposed catalytic mechanism for CDP-ME kinase as deduced from the structure determination described in the text. |
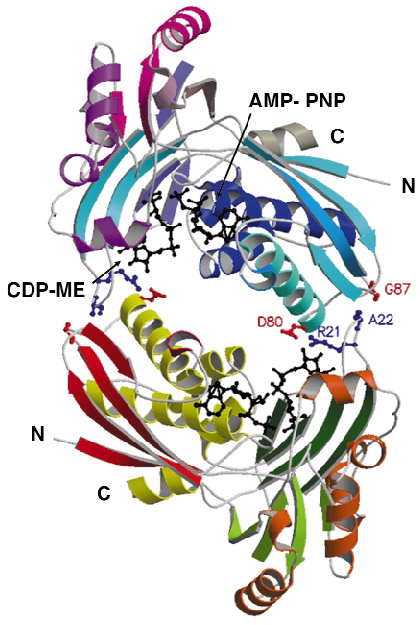
Although the enzyme crystallised readily, the crystals were poorly ordered and the ESRF facilities were essential to obtain high-quality diffraction data. After testing a great many samples, the structure of a ternary complex with substrate and an ATP analogue was determined at 2.0 Å resolution using the single-wavelength anomalous dispersion method. The asymmetric unit of the crystals contains two copies of the enzyme, which form an extended homodimer with C2 symmetry (Figure 29). The structure of the enzyme itself is characteristic of the Galacto-Homoserine-Mevalonate-Phosphomevalonate kinase superfamily, and consists of two domains, one of which binds the co-factor and one of which binds the substrate. The catalytic center is positioned in a deep cleft between domains. The cofactor ATP binds with its adenine base in an aliphatic cleft where it forms hydrogen-bonding interactions that stabilise the uncommon syn orientation of the base. Both the ribose and phosphate groups of the cofactor are solvent accessible. CDP-ME binds with the cytosine tucked inwards and sandwiched between two aromatic side chains. Its tail is directed towards the cofactor ![]() -phosphate at the catalytic center, which consists of Lys10 and Asp141 (Figure 28b). These residues polarise the O2M hydroxyl group of CDP-ME and facilitate proton abstraction. Asp141 acts as a general base and generates a nucleophile that attacks the ATP
-phosphate at the catalytic center, which consists of Lys10 and Asp141 (Figure 28b). These residues polarise the O2M hydroxyl group of CDP-ME and facilitate proton abstraction. Asp141 acts as a general base and generates a nucleophile that attacks the ATP ![]() -phosphate group while Lys10 stabilises the transition state.
-phosphate group while Lys10 stabilises the transition state.
 |
|
Fig. 29: Ribbon diagram representation of the structure of the CDP-ME kinase dimer. Amino acid residues contributing to dimer formation are shown as coloured balls and sticks while both the substrate and cofactor analogue are shown as black balls and sticks. |
The sequence identity of CDP-ME kinase averages 45% over all species with conservation extending throughout the sequence. In particular the key residues for ligand binding and catalysis are conserved. This high level of sequence similarity indicates that our model of the structure of CDP-ME kinase provides an excellent high-resolution template for a structure-based approach to aid the search for novel broad-spectrum antimicrobial drugs. One of the keys to the success of this endeavor will be the continued use of synchrotron radiation to ensure that accurate structural information for potential enzyme-drug complexes is attained.
Reference
[1] F. Rohdich, S. Hecht, A. Bacher and W. Eisenreich, Pure Appl. Chem. 75, 393-405 (2003).
Principal Publication and Authors:
L.Miallau (a,b), M.S. Alphey (a), L.E. Kemp (a), G.A. Leonard (b), S.M. McSweeney (b),S. Hecht (c), A. Bacher (c), W. Eisenreich (c), F. Rohdich (c) and W.N. Hunter (a), Proc. Natl. Acad. Sci. USA 100, 9173-9178 (2003).
(a) School of Life Sciences, University of Dundee (UK)
(b) ESRF
(c) Institute für Organische Chemie und Biochemie, Technische Universität München (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.