- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2002
- Macromolecular Crystallography
- Crystal Structure Reveals Blood Platelet Adhesion Complex
Crystal Structure Reveals Blood Platelet Adhesion Complex
Integrity of the blood circulation is crucial to survival. Therefore, blood loss from damaged vessels should be stopped rapidly. An essential first step in the arrest of bleeding is the adhesion of blood platelets to sites of vascular damage. In rapidly-flowing blood, this depends on the interaction between the glycoprotein Ib (GpIb
(GpIb![]() ), located at the platelet surface, and the multimeric blood protein von Willebrand Factor (VWF) immobilised at sites of vessel damage. The importance of the interaction between GpIb
), located at the platelet surface, and the multimeric blood protein von Willebrand Factor (VWF) immobilised at sites of vessel damage. The importance of the interaction between GpIb![]() and VWF is highlighted by the familial bleeding disorders von Willebrand's disease and the Bernard-Soulier syndrome that are caused by mutations in GpIb
and VWF is highlighted by the familial bleeding disorders von Willebrand's disease and the Bernard-Soulier syndrome that are caused by mutations in GpIb![]() and VWF. The interaction between GpIb
and VWF. The interaction between GpIb![]() and VWF also plays a pivotal role in the onset of thrombosis, a condition in which undesired platelet-aggregates are formed that can block blood vessels causing heart- and brain-infarctions.
and VWF also plays a pivotal role in the onset of thrombosis, a condition in which undesired platelet-aggregates are formed that can block blood vessels causing heart- and brain-infarctions.
We used data sets collected at beamlines ID14-1 and ID14-2 to determine crystal structures of the VWF-binding domain of GpIb![]() and of its complex with the VWF-A1 domain at resolutions of 1.9 Å and 3.1 Å, respectively. For the structure determination of the complex we crystallised the mutant proteins A1(R543Q) and GpIb
and of its complex with the VWF-A1 domain at resolutions of 1.9 Å and 3.1 Å, respectively. For the structure determination of the complex we crystallised the mutant proteins A1(R543Q) and GpIb![]() (M239V). These mutations are related to bleeding disorders and enhance complex formation.
(M239V). These mutations are related to bleeding disorders and enhance complex formation.
 |
|
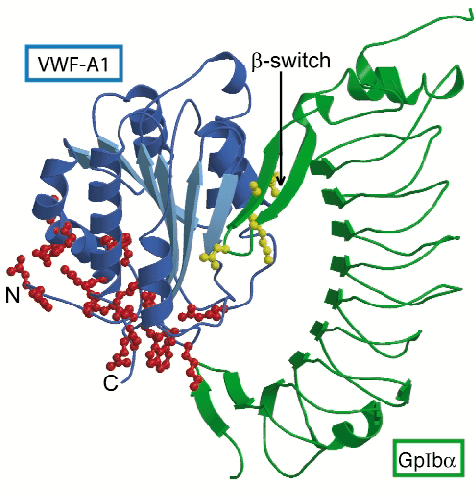
Fig. 5: Schematic representation of the complex of GpIb |
The VWF-binding domain of GpIba displays an elongated curved shape (Figure 5). The crystal structure of the complex shows that the globular A1 domain interacts with the concave face of GpIb![]() with two sites of close interaction. Interaction at the larger contact site near the top of the A1 domain depends on conformational changes in a surface-exposed loop of GpIb
with two sites of close interaction. Interaction at the larger contact site near the top of the A1 domain depends on conformational changes in a surface-exposed loop of GpIb![]() . This loop, which we call ß-switch, is flexible in free GpIb
. This loop, which we call ß-switch, is flexible in free GpIb![]() , whereas in the complex it forms a ß-hairpin that binds to the central ß-sheet of A1. Mutation M239V and in fact all other mutations in GpIb
, whereas in the complex it forms a ß-hairpin that binds to the central ß-sheet of A1. Mutation M239V and in fact all other mutations in GpIb![]() that cause stronger binding to VWF are located in the ß-switch. Because the activating mutations introduce residues known to stabilise ß-hairpin conformations (valine in the strand region or glycine in the turn region) enhanced complex formation probably results from stabilisation of the ß-hairpin conformation of the ß-switch, which primes GpIb
that cause stronger binding to VWF are located in the ß-switch. Because the activating mutations introduce residues known to stabilise ß-hairpin conformations (valine in the strand region or glycine in the turn region) enhanced complex formation probably results from stabilisation of the ß-hairpin conformation of the ß-switch, which primes GpIb![]() for binding to A1. The second, smaller contact site, located at the base of A1 appears to require the dislodging of the termini of the A1 domain uncovering the site of interaction. Mutations in A1 that cause increased binding to GpIb
for binding to A1. The second, smaller contact site, located at the base of A1 appears to require the dislodging of the termini of the A1 domain uncovering the site of interaction. Mutations in A1 that cause increased binding to GpIb![]() are all located at the base of A1 and probably induce the uncovering of the minor binding site by destabilising the conformation of its termini.
are all located at the base of A1 and probably induce the uncovering of the minor binding site by destabilising the conformation of its termini.
We propose that dislodging of the termini of the A1 domain under physiological conditions may be caused by pulling forces exerted on the immobilised VWF molecule by the rapidly flowing blood, thus providing activation in the platelet adhesion process. The two sites of interaction between GpIb![]() and VWF-A1 identified in this study present primary targets for development of drugs for the treatment or prevention of thrombosis.
and VWF-A1 identified in this study present primary targets for development of drugs for the treatment or prevention of thrombosis.
Principal Publication and Authors
E.G. Huizinga (a), S. Tsuji (b), R.A.P. Romijn (b), M.E. Schiphorst (b), Ph.G. de Groot (b), J.J. Sixma (b) and P. Gros (a), Science 297, 1176-1179 (2002).
(a) Crystal and Structural Chemistry, Utrecht University (The Netherlands)
(b) Department of Haematology, University Medical Center Utrecht (The Netherlands)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.