- Home
- News
- Spotlight on Science
- Working out the...
Working out the fundamental processes at metal-organic interfaces
23-05-2008
Electronic and optoelectronic devices based on organic semiconductors are currently entering the consumer electronics market. Nevertheless, many fundamental properties of their components have yet to be discovered. In particular, the development of an in-depth understanding of the detailed electronic structure of the interface between the charge injecting electrodes and the active organic semiconducting material is a topic of intense multi-disciplinary research.
It has recently been shown that interfacial (sub-) monolayers of strong electron acceptor molecules can be used to tune charge carrier injection barriers [1]. Based on UV-photoemission measurements it had been argued that the metal work-function increase that can be realised with these layers is due to a charge transfer from the metal to the lowest unoccupied molecular orbital (LUMO). However, by employing quantum-mechanical calculations in combination with X-ray standing wave (XSW) and photo-electron spectroscopy (XPS and UPS) experiments, it was shown that the actual situation is more complex for the prototypical case of the strong acceptor molecule F4TCNQ (2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane) adsorbed on the Cu(111) surface.
 |
|
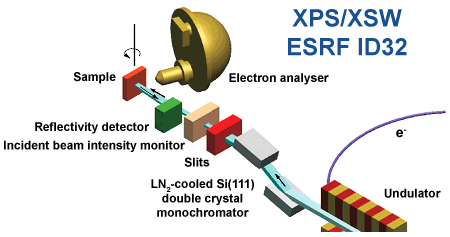
Figure 1. Schematic diagram of the X-ray standing wave setup at ID32. |
The experimentally-challenging XSW measurements were performed at ID32 [2,3], Figure 1 presents the experimental layout. The measurements revealed that the adsorption geometry of F4TCNQ on Cu deviates strongly from the planar configuration of the free molecule. The bonding distances of the different atoms within the organic molecule are 2.7±0.1 Å for the nitrogen and 3.3±0.1 Å for the fluorine atoms (Figure 2). Using these experimental values as reference for quantum-mechanical calculations one can uncover the electronic structure of the adsorbate complex: the strong interaction between F4TCNQ and Cu results in significant hybridisation between molecular and metallic states. Indeed, as verified by UPS measurements and by calculations, nearly all molecular LUMO derived states become occupied by electrons. This alone would give rise to an interface dipole of several eV, however, this is not realistic and here the calculations allow the complex nature of the charge transfers occurring upon adsorption to be tracked down. The filling of the LUMO is counteracted by an electron donation from deep lying σ-states localised on the –CN groups of the molecule back into the metal. Moreover, both the XSW data as well as the calculations indicate that the molecule becomes significantly distorted upon bonding to the metal (Figure 3) – an effect that we have also observed for several other strongly interacting molecular species [2,3]. This provides the otherwise centro-symmetric molecule with a significant dipole moment perpendicular to the metal surface, which gives rise to another shift of the vacuum level above the sample surface counteracting the results of the charge rearrangements between the metal and the molecule (Figure 4). In analogy to the situation encountered in covalently bound self-assembled monolayers of donor/acceptor substituted molecules [4], the overall modification of the metal work-function can be separated into two contributions, one arising from bond formation and another stemming from the molecular dipole moment. The conceptual difference is that for molecules like F4TCNQ, not only the bonding but also the “molecular” dipole moment is a consequence of the interaction with the substrate. The actual electronic structure of the interface results from a delicate balance between both effects.
In conclusion, it was demonstrated that the entangled electronic and structural properties of organic molecules on surfaces require a sophisticated experimental and theoretical approach. In this context the XSW measurements performed at the ESRF provide very valuable and precise information which cannot easily be obtained by other means.
 |
|
Figure 2. X-ray standing wave data measured for F4TCNQ on Cu(111). |
 |
|
Figure 3. Calculated adsorption geometry of F4TCNQ on Cu(111). |
 |
|
Figure 4. Charge rearrangements upon bond formation between F4TCNQ and the Cu(111) surface. Electrons are shifted from the blue to the red areas. |
References
[1] N. Koch, S. Duhm, J.P. Rabe, A. Vollmer, and R.L. Johnson, Phys. Rev. Lett. 95, 237601 (2005).
[2] A. Gerlach, S. Sellner, F. Schreiber, N. Koch, and J. Zegenhagen, Phys. Rev. B 75, 045401 (2007).
[3] A. Gerlach, F. Schreiber, S. Sellner, H. Dosch, I.A. Vartanyants, B.C.C. Cowie, T.-L. Lee, and J. Zegenhagen, Phys. Rev. B 71 205425 (2005).
[4] G. Heimel, L. Romaner, E. Zojer, and J.L. Brédas, Nano Lett. 7, 932 (2007).
Principle publication and authors
L. Romaner (a), G. Heimel (b), J.L. Brédas (b), A. Gerlach (c), F. Schreiber (c), R.L. Johnson (d), J. Zegenhagen (e), S. Duhm (f), N. Koch (f), and E. Zojer (a), Phys. Rev. Lett. 99, 256801 (2007).
(a) Institut für Festkörperphysik, Technische Universität Graz (Austria)
(b) School of Chemistry and Biochemistry, Georgia Institute of Technology (USA)
(c) Institut für Angewandte Physik, Universität Tübingen, (Germany)
(d) Institut für Experimentalphysik, Universität Hamburg (Germany)
(e) ESRF
(f) Institut für Physik, Humboldt-Universität zu Berlin (Germany)
What's related?
Beamline ID32
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.