- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2010
- Soft condensed matter
- Ultrafast X-ray solution scattering reveals different reaction pathways in the photolysis of Ru3(CO)12 after ultraviolet and visible excitation
Ultrafast X-ray solution scattering reveals different reaction pathways in the photolysis of Ru3(CO)12 after ultraviolet and visible excitation
The triangular metal carbonyl cluster, Ru3(CO)12, plays an important role in photocatalysis and photoenergy conversion and has served as the paradigm for the photochemistry of transition metal carbonyls. However, detailed mechanisms for the photochemical reactions are rarely available, mainly due to the lack of efficient methods to study them. Ultrafast X-ray solution scattering has been shown to give information that is generally difficult to extract from ultrafast optical spectroscopy such as the time course of changes in bond lengths and angles, including those of short-lived intermediates, on a timescale of picoseconds to milliseconds [1]. Following this approach, our recent time-resolved X-ray solution scattering study on Ru3(CO)12 in cyclohexane excited at 390 nm revealed a new intermediate, Ru3(CO)10, that escaped detection in a previous spectroscopic study [2].
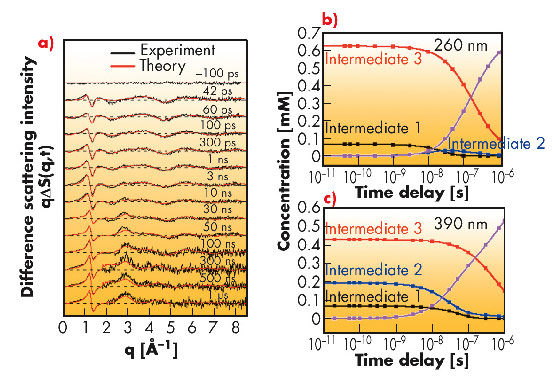
The UV-vis spectrum of Ru3(CO)12 in cyclohexane has two prominent absorption bands: the first centered at 390 nm and the second with a peak at 238 nm and an absorption shoulder at 260 nm. Electronic absorption studies indicate that the lower energy band at 390 nm originates from the electronic transition σ→σ* in metal d-orbitals, resulting in heterolytic cleavage of one of the Ru-Ru bonds. The short-wavelength absorption band in the UV range has been attributed to metal to ligand charge transfer (MLCT) which ultimately results in loss of one carbonyl group in solution. Ultrafast X-ray solution scattering illustrated that these different absorption processes lead to distinct photofragmentation pathways when Ru3(CO)12 is excited by different wavelengths. The photodissociation of Ru3(CO)12 dissolved in cyclohexane was studied on beamline ID09B employing a pump-probe experimental setup. Ultraviolet (260 nm) and visible (390 nm) laser pulses (2 ps) were used for excitation and 100 picosecond X-ray pulses for probing the transient intermediates. In these experiments, the sample flows through a nozzle that produces a layer of liquid of 300 µm thickness. The pump-probe sequence is repeated with different time delays between pump and probe at a frequency of 986.3 Hz and the scattered signal is accumulated on a MarCCD detector. The difference X-ray scattering intensities (qΔS(q,t)) illustrating the structural changes due to the laser excitation are shown in Figure 70a as a function of different time delays.
 |
|
Fig. 70: a) Time-resolved difference scattering intensities qΔS(q,t) as a function of time delay after photolysis of Ru3(CO)12 in cyclohexane at 260 nm. The black curves correspond to the experimental data and the red curves to the least-squares fits. Comparison of the time course of the concentration changes of intermediates 1 (black), 2 (blue) and 3 (red) during the photoreaction of Ru3(CO)12, after excitation b) with 260 nm and c) with 390 nm. |
Time-resolved X-ray solution scattering on photolysis of Ru3(CO)12 in cyclohexane at 260 nm and 390 nm reveals different photodissociation pathways. Upon UV excitation at 260 nm, at the onset of the reaction, the species formed are only Ru3(CO)11(µ-CO) for the metal-metal cleavage channel and the intermediate Ru3(CO)10 with 2 CO loss. In the course of the reaction, the major photoproduct Ru3(CO)10 then recombines with a free CO to Ru3(CO)10(µ-CO), which eventually decays into the starting molecule Ru3(CO)12 by recombination with another CO (Figure 70b). After excitation at 390 nm, three intermediates are formed from the initial molecule Ru3(CO)12 at the onset of the reaction: Ru3(CO)11(µ-CO), Ru3(CO)10(µ-CO) with bridged CO and Ru3(CO)10 with terminal CO only (Figure 70c). The different photofragmentation pathways of Ru3(CO)12 in cyclohexane upon UV excitation presumably originate from the higher photon energy which favours the simultaneous loss of two CO leading to Ru3(CO)10 only for the CO loss reaction channel at the onset of reaction (Figure 71).
 |
|
Fig. 71: Ultrafast X-ray solution scattering reveals different reaction pathways in the photolysis of Ru3(CO)12 at 260 nm and 390 nm. The major difference in the photolysis at 260 nm and 390 nm is due to Ru3(CO)10(µ-CO). After excitation at 260 nm, it is formed from Ru3(CO)10 by recombination with a free CO, whereas, after excitation at 390 nm, it is formed from the starting molecule Ru3(CO)12 at the onset of the reaction. The commonality of the reaction between 260 nm and 390 nm excitation is the formation of Ru3(CO)10 as the major photoproduct. In the figure, the solid arrows show the reaction pathways at 260 nm, while solid plus dash arrows show the reaction pathways at 390 nm. |
Principal publication and authors
Q.Y. Kong (a), J.H. Lee (b), K.H. Kim (b), J.H. Kim (b), M. Wulff (c), H. Ihee (b) and M.H.J. Koch (d), J. Am. Chem. Soc. 132, 2600-2607 (2010).
(a) Synchrotron SOLEIL, GIF-sur-Yvette (France)
(b) Center for Time-Resolved Diffraction, Department of Chemistry, Graduate School of Nanoscience and Technology (WCU), KAIST, Daejeon (Republic of Korea)
(c) ESRF
(d) EMBL, Hamburg Outstation,EMBL c/o DESY, Hamburg (Germany)
References
[1] T.K. Kim, J.H. Lee, M. Wulff, Q.Y. Kong and H. Ihee, ChemPhysChem 10, 1958-1980 (2009).
[2] Q.Y. Kong, J.H. Lee, A. Plech, M. Wulff, H. Ihee and M.H.J. Koch, Angew. Chem., Int. Ed. 47, 5550-5553 (2008).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.