- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Structural biology
- The structural basis for ion pumping by integral membrane pyrophosphatases
The structural basis for ion pumping by integral membrane pyrophosphatases
Plants, parasitic protozoa, bacteria and archae contain membrane-integral pyrophosphatases (M-PPases). These are novel primary transmembrane pumps with 14-17 transmembrane (TM) helices that link pyrophosphate (PPi) hydrolysis to sodium or proton pumping [1]. PPases are essential, they make it possible for anabolic reactions like DNA synthesis to be driven to completion. Unlike the soluble PPases, M-PPases recycle part of the free energy of PPi hydrolysis to generate an electrochemical potential across biological membranes. In plants, they are vital for maturation and enhance survival under abiotic stress conditions such as drought, anoxia and cold [2]. They are also important for proliferation of disease causing protozoa [3].
M-PPases have neither sequence, structural nor functional similarity to the FoF1 or P-type ATPases. They can either be proton or sodium pumps, and some require K+ for maximal activity. The resting enzyme state is EMg2, and two more metal ions bind with substrate, Mg2PPi [2].
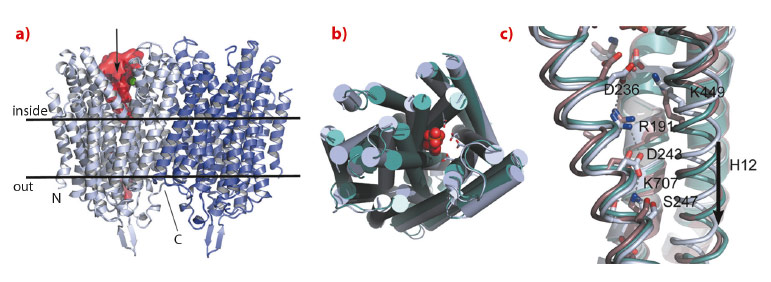
We have solved the structure of the Na+-pumping M-PPase of Thermotoga maritima (TmPPase) in the resting state (TmPPase:Ca:Mg) at 2.6 Å resolution and with product bound (TmPPase:Pi2:Mg42) at 4.0 Å. TmPPase is a homodimer with 16 TM-helices per monomer (Figure 7). The helices extend up to 25 Å from the membrane bilayer on the cytoplasmic side so that the hydrolytic centre is held some 20 Å above the membrane plane.
 |
|
Fig. 7: Structure of Thermotoga maritima membrane bound pyrophosphatase and a comparison of different catalytic states. a) The dimer structure of Tm-PPase in relation to the lipid bilayer: one active site is indicated by the green metal ion (Mg2+), the cavity space in red, with an arrow indicating substrate binding. b) Comparison of the apo-structure (light blue) and Pi-product complex (active site viewed from above) of Tm-PPase (green), showing the contraction of the structure around the active site with bound ligand (2 x Pi2, shown as red spheres). c) Comparison of the Tm-PPase apo-structure (light blue), product complex (green), and mung bean PPase inhibitor complex (brown) around the gate region, showing the salt bridges involving R191 and the downward movement of TM-helix 12 (a and c, adapted from Kellosalo et al. 2012). |
The protein has a completely novel fold and its unusual active site has four distinct regions: the hydrolytic centre, a “coupling funnel”, the gate (closed in our structure) just below the membrane surface, and an exit channel for Na+-ions. The distance from the hydrolytic centre to the gate is about 20 Å. Six helices (5-6, 11-12 and 15-16) form the hydrolytic centre and the coupling funnel on the cytoplasmic face of the protein. However, just four helices (5, 6, 12 and 16) form the gate and channel, creating an internal “symmetry mismatch” between the two sides of the protein.
Below the hydrolytic centre is the “coupling funnel” (Figure 7) containing eight absolutely conserved charged residues that couple hydrolysis to ion pumping. Some of the interactions observed in the resting structure suggest that the funnel is poised to switch into an alternate conformation to pump the sodium ion, especially the residue R191, which is almost equidistant between D243 and D236. Alignment of our structures and that of the substrate complex of the related mung bean PPase [4] shows that TM12 moves downwards by about 2 Å, concomitant with R191 switching up and down in a locked and released spring movement.
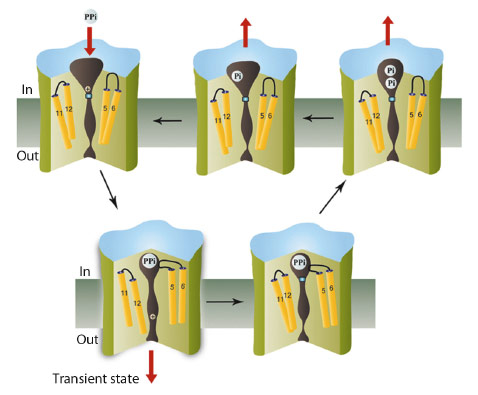
As the conformational changes observed in the inhibited and product-bound states are similar, with the exit channel closed in both, we suggest that ion pumping in M-PPases occurs via a “binding change” mechanism as in the FoF1 ATP synthase, and that the full conformational change that opens the gate involves a downwards piston-like motion of TM12 (Figure 8). This is driven by a concerted contraction during substrate binding. This change destroys a high-affinity sodium-binding site above the gate, leading to transfer of the ion into the exit channel, followed by its rapid diffusion into the periplasm. Consistent with this model, there are no conserved polar residues below the gate. Release of the sodium ion would allow the gate to close, hydrolysis to occur, product to leave, and the protein to revert to its resting state.
 |
|
Fig. 8: Schematic showing the catalytic cycle of membrane pyrophosphatases, with the transition state indicated. Three of the accessible states have been solved by us, while the substrate-analogue complex (lower right) was solved by Lin et al. [4]. |
Our work and the related mung bean structure provide the first steps in understanding the structure and mechanism in this novel family of ion pumps.
Principal publication and authors
J. Kellosalo (a), T. Kajander (a), K. Kogan (a), K. Pokharel (a,b) and A. Goldman (a), Science 337 473-476 (2012).
(a) Structural Biology and Biophysics Program, Institute of Biotechnology, University of Helsinki (Finland)
(b) Department of Biochemistry and Food Chemistry, University of Turku (Finland)
References
[1] A.M. Malinen, G.A. Belogurov, A.A. Baykov and R. Lahti, Biochemistry 46, 8872–8878 (2007).
[2] M. Maeshima, Biochim. Biophys. Acta 1465, 37–51(2000).
[3] M.T. McIntosh and A.B. Vaidya, Int. J. Parasitol. 32, 1–14 (2002).
[4] S.M. Lin, J.Y. Tsai, C.D. Hsiao, Y.T. Huang, C.L. Chiu, M.H. Liu, J.Y. Tung, T.H. Liu, R.L. Pan and Y.J. Sun, Nature 484, 399–403 (2012).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.