- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Structural biology
- Ligand screening and in situ synthesis using protein crystals
Ligand screening and in situ synthesis using protein crystals
Drug discovery is an active field of research in which medicinal chemistry remains a key player and X-ray crystallography is of increasing importance. Indeed, crystal structures of protein targets bound to ligands are providing essential guidelines for chemical structure optimisation. X-ray crystallography can also be used to screen for so-called fragments, i.e. weak and small binders, that can be built up step-by-step to produce larger and better ligands [1]. In parallel, organic chemistry has witnessed the emergence of soft reactions which work efficiently at room temperature (and pressure) and in aqueous solvent making them a priori compatible with biological samples. Such biocompatible chemical reactions have been used to link together fragments taking advantage of the protein target to template ligand growth [2]. However, this approach has been limited by the size of the combinatorial libraries one can deconvolute after in situ reaction, and by the number of biocompatible reactions. Surprisingly, structure-based fragment screening and in situ chemistry have not been combined extensively, despite a clear complementarity and proof-of-concept [3]. Moreover, molecular targets of drugs have long been viewed as static and inert structures and not as potential catalysts of new in situ reactions despite a seminal example of so-called epitaxial selection [4].
In this context, we were interested in studying bacterial NAD kinases that are essential enzymes and represent attractive therapeutic targets for the development of new antibiotics. So far, little knowledge has been gathered for these proteins as their coding genes have only recently been discovered and no inhibitor was known. In order to rapidly obtain new ligands, we carried out focused screening using adenosine derivatives as mimics of the two nucleotidic moieties of NAD, the natural substrate. Our expectation was to map the active site and to introduce a chemical ‘handle’ to which we could tether additional fragments. Accordingly, some adenosine congeners were designed, synthesised, and then co-crystallised with the enzyme. Among the seven 5’-adenosine derivatives tested, only two gave rise to well-diffracting crystals while crystal soaking added two new structures (recorded at beamlines ID14-4 and ID14-2 at resolutions between 1.75 Å and 2.03 Å). The refined structures showed perfect mimicry of the biologically relevant substrate NAD, and revealed a central hydrophobic sub-pocket. This suggested that the diphosphate moiety of NAD could be substituted by a short, rather hydrophobic linker between two adenosines in order to design a novel inhibitor.
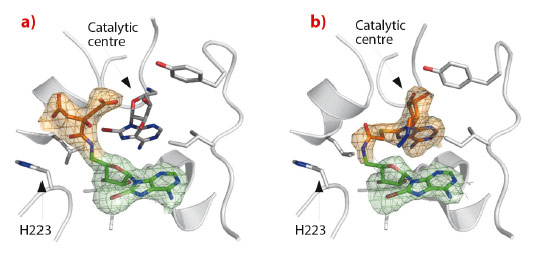
Subsequently, we analysed the binding of 8-bromo-adenosines and discovered an unanticipated amidation catalysed by NAD kinase. Soaking the crystals with a new compound (5’-amino-8-bromo-5’-deoxyadenosine) revealed extra electron density corresponding to a citrate molecule (a crystallisation agent) attached covalently to the bound adenosine (Figure 5a). According to the clear electron density, an amide bond was formed. This chemical reaction is distinct from the biologically relevant activity of this enzyme, and occurs at a position 7 Å away from the actual catalytic centre. This unexpected reactivity prompted us to bridge two adenosine derivatives in situ in order to completely fill the protein active site (Figure 5b). Then, a closely related compound with no labile bromide atom was synthesised chemically, and was shown to inhibit in vitro the growth of the human pathogen Staphylococcus aureus.
 |
|
Fig. 5: In situ amidation. Crystal structures of NAD kinase in complex with two products of in situ chemistry: a) 8-bromo-5’-amino-5’-deoxyadenosine (green) with covalently linked citrate (orange); b) 8-bromo-5’-amino-5’-deoxyadenosine (green) covalently linked in situ with 8-thioglycolic-5’-azido-5’-deoxyadenosine (orange). Data recorded on ID14-2 and ID14-4 at resolutions of 2.38 Å and 2.29 Å respectively. |
Interestingly, the in situ formation of the amide bond is reminiscent of the removal of the 5’-thioacetyl group of another adenosine derivative by the same enzyme [5]. This led to a model where a nearby histidine (H223), normally interacting with a phosphate group of the natural ligand, stabilises the tetragonal intermediate of the amidation and deacetylation reactions. Indeed, mutation of the histidine abolishes the amidation confirming the role of the protein in the reaction.
Structure-based drug design is a promising method but its combination with in situ chemistry is still in its infancy. Here, we have shown the possible use of a protein to build up its own ligands following focused ligand screening by X-ray crystallography. This approach takes advantage of the power of high-quality beamlines and automated crystal handling that allow rapid and efficient crystal screening (100-200 datasets collected per day).
Principal publication and authors
M. Gelin (a,b), G. Poncet-Montange (a,b), L. Assairi (c,d), L. Morellato (e,f), V. Huteau (e,f), L. Dugué (e,f), O. Dussurget (g,h,i), S. Pochet (e,f) and G. Labesse (a,b), Structure 20, 1107–1117 (2012).
(a) CNRS, UMR5048, Universités Montpellier 1 et 2; Centre de Biochimie Structurale, Montpellier (France)
(b) INSERM, U1054, Montpellier (France)
(c) Institut Curie, Orsay (France)
(d) INSERM, U759, Orsay (France)
(e) Unité de Chimie et Biocatalyse, Institut Pasteur, Paris (France)
(f) CNRS, UMR3523, Paris (France)
(g) Unité des Interactions Bactéries-Cellules, Institut Pasteur, Paris (France)
(h) INSERM U604, Paris (France)
(i) INRA USC2020, Paris (France)
References
[1] G. Chessari and A.J. Woodhead, Drug Discov Today. 14, 668-675 (2009).
[2] M.F. Schmidt and J. Rademann, Trends Biotechnol. 27, 512-521 (2009).
[3] M.S. Congreve et al., Angew Chem Int Ed Engl. 42, 4479-4482 (2003).
[4] B.A. Katz et al., Biochemistry 34, 8264-8280 (1995).
[5] G. Poncet-Montange, et al., J. Biol. Chem. 282, 33925-33934 (2007).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.