- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2012
- Structure of materials
- Smooth plating: Surface diffraction studies detect anomalous growth of copper
Smooth plating: Surface diffraction studies detect anomalous growth of copper
Copper galvanic plating is a widely used industrial process with numerous applications. Because of the nanoscale dimensions of current copper interconnects, precise control of the electrochemical deposition processes, and hence an improved fundamental understanding of the underlying elementary steps, is required. In contrast to Cu homoepitaxy under UHV conditions, fundamental understanding of the electrodeposition of Cu is largely lacking. Specifically, the precise influence of the solid-liquid interface structure and the applied electrode potential on the elementary surface processes involved in electrodeposition is unclear.
The presence of small amounts of chloride in the acidic plating bath is known to be vital to obtain optimal growth behaviour and deposition properties in almost any industrial plating process. However, how this simple inorganic species influences the Cu growth on the atomic scale is surprisingly still unresolved. To obtain deeper insight into these processes, homoepitaxial electrochemical growth of Cu(001) in a chloride-containing acidic solution was studied in operando by surface X-ray diffraction at beamline ID32 (Eγ = 22.5 keV).
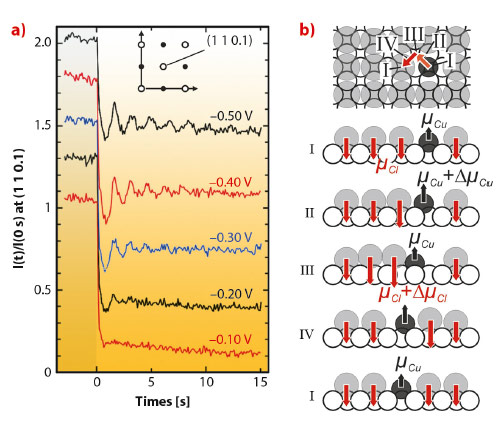
The experiments were performed using an electrochemical three-electrode cell (“hanging meniscus” cell), where the electrode is contacted by a droplet of the electrolyte. The high-speed surface scattering setup allowed the Cu growth to be studied at rates of several monolayers per second by recording the time-dependent intensity of the crystal truncation rod close to the anti-Bragg position. The Cu-sample potential was first kept at –0.6 VAg/AgCl, where the high Cu deposition rate and very high surface mobility result in a rapid smoothening even of rough surfaces. Then the potential was gradually increased. Analogous to MBE growth studies, different kinetic growth modes result in characteristic intensity transients at various constant deposition rates (Figure 132a). Characteristic intensity oscillations indicate layer-by-layer growth in an intermediate potential regime, whereas crossover to 3D growth is observed at the most positive potentials.
 |
|
Fig. 132: a) Scattered intensity I(t) at (1 1 0.1) during Cu electrodeposition from 0.1 M HClO4 + 1 mM HCl + 5 mM Cu(ClO4)2 on Cu(001) at different potentials. b) Illustration of Cu adatom diffusion on Cu(001) / c(2×2) Cl, arrows in I to IV indicate surface dipole moments of Cu (µCu) and Cl (µCl) adsorbates. |
Thus, the Cu surface mobility is reduced at positive electrode potential. This is in stark contrast to the potential dependence commonly found for metal self-diffusion on electrode surfaces. Usually, higher mobility is found at more positive potentials as demonstrated by our previous results on the homoepitaxial electrodeposition on Au(001) in Cl-containing electrolyte [1] and is also predicted by the current theory of surface transport at electrochemical interfaces [2]. It is explained by an electrostatic energy contribution, resulting from the interaction of the metal adatom’s dipole moment with the strong electric field of the electrochemical double layer.
The surprising, anomalous behaviour of Cu(001) can be explained by including the presence of the co-adsorbed c(2×2) chloride adlayer into this theory. Apparently, it strongly modifies and actually inverts the effective change in the dipole moment during the metal adatom hopping. As schematically illustrated in Figure 132b, the metal adatoms have to move through this rather close-packed adlayer, displacing neighbouring Cl from their preferred fourfold-hollow adsorption sites. The resulting additional dipole contribution of those rather anionic species is opposite in sign to that of the metal adatom itself and can dominate the potential-dependent term of the surface diffusion barrier. In other words, Cu atoms moving across the surface have to push through the Cl layer and become more mobile at more negative electrode potentials, where the chloride is more weakly bound. On Au(001) this coadsorbate contribution is smaller, probably due to differences in the adlayer structure and charge state of the Cl adsorbates.
Our findings not only aid substantially the fundamental understanding of growth and surface transport in electrochemical environments, but also have significant practical implications. Strongly chemisorbing anions such as halides are ubiquitously present in technological electrodeposition processes. Knowledge on whether the electrode surface transport is normal or anomalous will allow an assessment of whether a lower roughness of the deposit may be expected at low or at high deposition overpotentials, respectively. The novel concept of an effective diffusion dipole moment provides a simple conceptual framework that allows microscopic physical growth theories to be extended to liquid phase deposition processes. Since these dipole moments can be obtained by ab initio calculations, our results open the way for predicting and quantifying the growth behaviour in these complex systems.
Principal publication and authors
F. Golks (a,b), J. Stettner (a), Y. Gründer (a), K. Krug (a), J. Zegenhagen (b) and O.M. Magnussen (a), Phys. Rev. Lett. 108, 256101-1 256101-5 (2012).
(a) Institut für Experimentelle und Angewandte Physik, Universität Kiel (Germany)
(b) ESRF
References
[1] K. Krug, J. Stettner and O.M. Magnussen, Phys. Rev. Lett. 96, 246101 (2006).
[2] M.Giesen et al., Surf. Sci. 595, 127 (2005).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.