- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2011
- Structural biology
- Snapshot of a bacterial transporter in action
Snapshot of a bacterial transporter in action
The human urinary tract is one of the most common sites of bacterial infection and most of them are caused by uropathogenic Escherichia coli (UPEC). During the infection, UPEC adhere to host epithelial cells by using specific extracellular adhesive organelles termed pili. Despite a wealth of other structural and biochemical information, little is known about how pilus formation is orchestrated at the bacterial cell surface. We present here a crystal structure capturing the pilus assembly platform in the act of secreting its cognate substrate. This new breakthrough provides new insights into the molecular details of pilus assembly.
Uropathogenic Escherichia coli (UPEC) attach specifically to human bladder cells using surface structures known as type 1 pili. These are assembled by the so-called chaperone-usher (CU) pathway, one of the best-characterised secretion systems in Gram-negative bacteria [1]. Type 1 pili consist of many individual subunits that are polymerised at the outer membrane by a specific assembly platform, the usher. In the periplasm, each subunit is stabilised by a dedicated molecular chaperone, FimC, which protects it from aggregation. The chaperone:subunit complexes are then targeted differentially to the usher FimD, which catalyses their assembly. The first subunit to be engaged is FimH, the adhesin at the very tip of the pilus, and subsequent subunits are added one at a time from the base up.
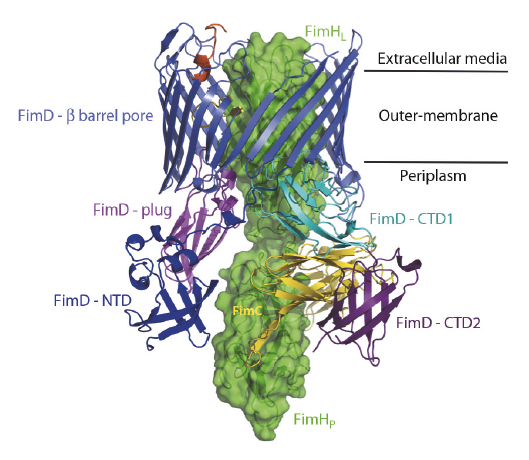
Over the last two decades, some of the molecular details of pilus biogenesis through the CU pathway have been provided [1]. However, little is known about how the usher works. We have recently solved a new crystal structure describing the first step in pilus biogenesis – the FimD usher bound to its cognate FimC:FimH substrate – using diffraction data collected at beamline ID23-1 (Figure 107). This structure, solved at 2.8 Å resolution, shows the full-length usher FimD in its active state. Along with the crystal structure of the closed state of the FimD pore domain, this structure sheds new light on the mechanism of usher activation.
 |
|
Fig. 107: Crystal structure of the FimD:FimC:FimH complex shown as side view and ribbon/surface representation. The adhesin subunit FimH is in green (FimHL and FimHp stand for the lectin and the pilin domains respectively) and the periplasmic chaperone FimC is in yellow. The usher FimD comprises 5 domains: the N-terminal domain (NTD), the C-terminal domains (CTD1 and CTD2), the plug domain and the β-barrel pore domain. The usher NTD, β-barrel, plug, CTD1 and CTD2 are in blue, slate, magenta, cyan and purple, respectively. Two black lines indicate the position of the usher within the bacterial outer-membrane. |
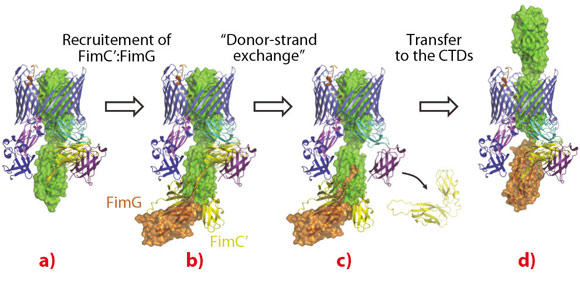
A striking feature of the active usher conformation is that the FimC:FimH complex is bound to a newly identified site on the C-terminal domains (CTDs) of FimD. This frees the N-terminal domain (NTD), which is known to bind chaperone-subunit complexes [2], to recruit the next subunit. Furthermore, this binding positions the two subunits in an ideal position for the polymerisation mechanism to take place (Figure 108). We thus conclude that the usher has two binding sites (NTD and CTDs) that act in concert, such that the growing pilus remains anchored to one, while the next subunit in assembly is recruited by the other (Figure 108).
 |
|
Fig. 108: Structural model of the subunit incorporation cycle at the FimD usher, represented as in Figure 107. a) The FimD:FimC:FimH complex. In the described conformation, the FimD NTD is in an ideal position for the recruitment of the next chaperone:subunit complex FimC’:FimG. b) FimC’:FimG (in yellow and orange, respectively) bound at the NTD of the usher (model based on PDB deposition 1ZE3 [2]). c) The FimG subunit polymerises with FimH via the “donor-strand exchange” mechanism, leading to the dissociation of FimC from FimH and from the usher CTDs. d) Transfer to the FimD CTDs and translocation of FimH through the FimD pore. This frees up the FimD NTD for the next round of subunit incorporation. |
Principal publication and authors
G. Phan (a), H. Remaut (a,b), T. Wang (c), W.J. Allen (a), K.F. Pirker (a), A. Lebedev (d), N.S. Henderson (e), S. Geibel (a), E. Volkan (f), J. Yan (a), M.B.A. Kunze (a), J.S. Pinkner (f), B. Ford (f,g), C.W. McKay (a), H. Li (c,h), S. Hultgren (f), D.G. Thanassi (e) and G. Waksman (a), Nature 474, 49-53 (2011).
(a) Institute of Structural and Molecular Biology, UCL and Birkbeck (UK)
(b) Structural Biology Brussels (Belgium)
(c) Biology Department, Brookhaven National Laboratory (USA)
(d) Department of Chemistry, University of York (UK)
(e) Center for Infectious Diseases, Stony Brook University (USA)
(f) Department of Molecular Microbiology and Center for Women’s Infectious Disease Research, Washington University School of Medicine (USA).
(g) Department of Pathology and Immunology, Washington University (USA)
(h) Department of Biochemistry and Cell Biology, Stony Brook University (USA)
References
[1] G. Waksman and S.J. Hultgren, Nature Review Microbiology 7, 765-767 (2009).
[2] M. Nishiyama et al., EMBO Journal 24, 2075-86 (2005).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.