- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2011
- Structural biology
- Structure of the membrane domain of respiratory complex I
Structure of the membrane domain of respiratory complex I
Mitochondria are “cellular power plants” supplying energy for the organism. They contain their own DNA, about half of which codes for proteins of mitochondrial complex I, the first and largest enzyme in the respiratory “power chain”. Defects in mitochondrial DNA are one of the most common types of human genetic disorder. They occur in about 1 in 5,000 of the population and the majority involve respiratory complex I. So far there is no treatment for the debilitating diseases resulting from these disorders, which include neurological impairment, deafness, blindness, muscle weakness and cardiovascular disease.
To understand the molecular basis of such diseases, as a starting point for drug development, we need to know the structure of Complex I. Complex I transfers two electrons from NADH to quinone and couples this process to the translocation of four protons across the membrane. It plays a central role in cellular energy production, providing about 40% of the proton flux required for ATP synthesis. Mitochondrial complex I consists of 45 subunits, whilst the prokaryotic enzyme is simpler, consisting of 14 “core” subunits with a total mass of about 550 kDa. The prokaryotic enzyme represents an important ‘minimal’ model of human complex I.
Complex I, due to its sheer size, resisted the efforts to determine its structure for a long time. Our earlier crystal structures of the hydrophilic domain of complex I from Thermus thermophilus established the electron transfer pathway from NADH through flavin mononucleotide (FMN) and seven iron-sulfur (Fe-S) clusters to the likely quinone binding site [1]. Recently we have determined the architecture of the entire complex at 4.5 Å resolution [2]. Now, we have succeeded in the determination of the crystal structure of the membrane domain, at 3.0 Å resolution. Most of the data was collected at beamline ID29. The membrane domain includes six subunits containing a total of 55 transmembrane (TM) helices and represents, to date, one of the largest membrane proteins for which the structure has been solved using X-ray crystallography.
The crystal structure reveals that the fold of three largest subunits (NuoL/M/N; homologous to Mrp antiporters) is novel, with two inverted structural repeats of five TM helices arranged, unusually, face-to-back. Each repeat includes a discontinuous TM helix, containing essential charged residues, and forms half of a channel across the membrane. A network of conserved polar residues connects the two half-channels, which completes the proton translocation pathway. Unexpectedly, lysines rather than carboxylate residues appear to act as the main elements of the proton pump in these subunits. The fourth likely proton-translocation channel is found at the interface of subunits NuoN/K/J/A and contains two conserved glutamates in the middle of the membrane.
 |
|
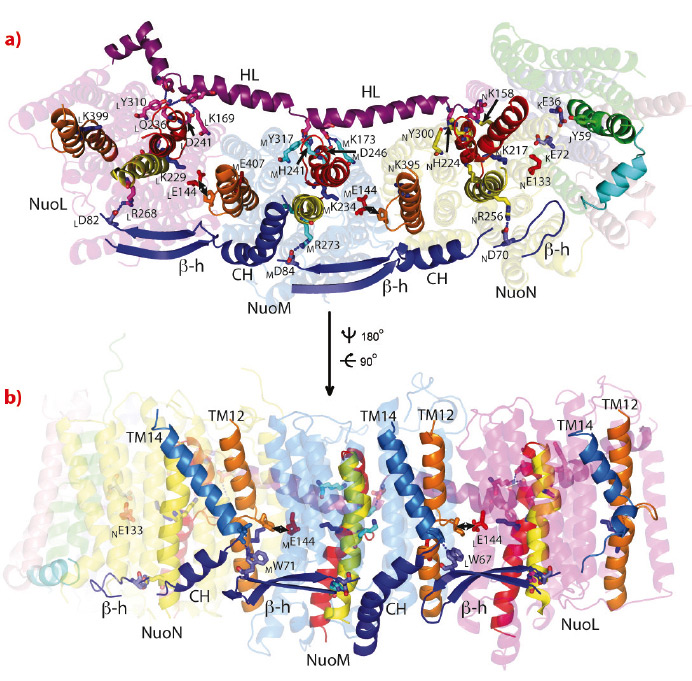
Fig. 113: Interactions between subunits of the membrane domain of Complex I. a) View from the periplasm; b) view in the membrane. Helix HL is in purple and the β-hairpin-helix element (β-h and CH) in blue. Helices likely involved in conformational changes are shown in red (LMNTM7), orange (TM12), yellow (TM8) and green (JTM3). Essential residues are shown as sticks and labelled (prefix indicates subunit). |
Antiporter-like subunits are connected linearly, like carriages in a train. Due to their internal symmetry, functionally important residues interact between the subunits, allowing communication of conformational changes (Figure 113). The subunits are held together on one side of the domain by an unusual long amphiphatic helix HL and on the other side by a βH motif consisting of connected in series β-hairpins and helices. Both structural elements interact with flexible discontinuous TM helices, containing essential lysine residues.
 |
|
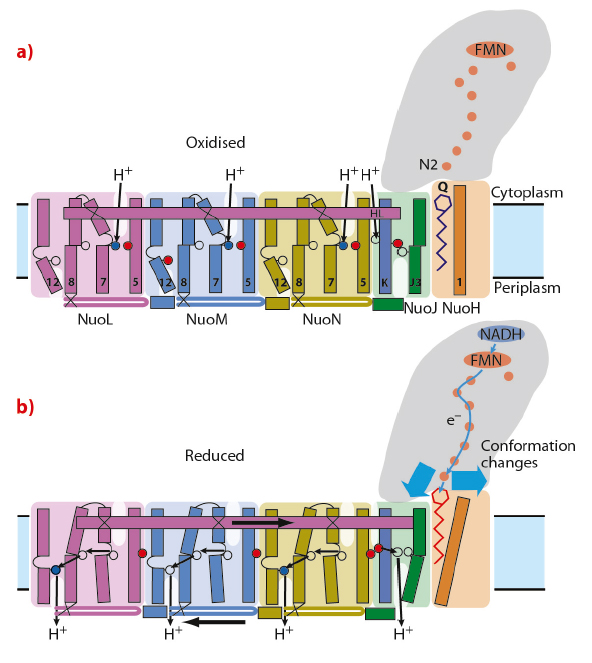
Fig. 114: Proposed mechanism of complex I. a) Oxidised state. b) Reduced state. Conformational coupling between electron transfer and proton translocation is mediated by helix HL (cytoplasmic side) and the βH element (periplasmic side). TM helices are numbered. Crucial charged residues are indicated by red/empty circles (Glu) and empty/blue circles (Lys) for unprotonated/protonated residue, respectively. In NuoL/M/N, LysTM7 from the first half-channel is assumed to be protonated in the oxidised state. Upon reduction it donates its proton to the connecting Lys/HisTM8 and then onto Lys/GluTM12 from the second half-channel. Lys/GluTM12 ejects its proton into periplasm upon return from reduced to oxidised state. A fourth proton per cycle is translocated at the interface of NuoN, K and J. |
Thus, the crystal structure shows that the enzyme functions by connecting different parts of the protein through mechanical coupling elements, akin to the piston coupling rods in a steam engine (Figure 114). The crystal structure also elucidates, for the first time, how the most common mutations in the membrane subunits lead to human disorders. The mutations are found in areas of the protein critically important for energy transduction. This structural knowledge should greatly help scientists developing drugs against mitochondrial diseases.
Principal publication and authors
R.G. Efremov (a,b) and L.A. Sazanov (a), Nature 476, 414–420 (2011).
(a) Medical Research Council Mitochondrial Biology Unit, Cambridge (UK)
(b) Present address: Max-Planck Institute for Molecular Physiology, Dortmund (Germany)
References
[1] L.A. Sazanov and P. Hinchliffe, Science 311, 1430-1436 (2006).
[2] R.G. Efremov, R. Baradaran and L.A. Sazanov, Nature 465, 441-445 (2010).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.