- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2009
- Soft condensed matter
- Pathways of micellar self-assembly probed by time-resolved SAXS
Pathways of micellar self-assembly probed by time-resolved SAXS
In material science and physical chemistry, self-assembly is an important route for manipulation and control for a rational design of nanostructures. Synthetic amphiphilic block copolymers belong to the family of self-assembling systems which, apart from the spontaneous self-assembly property, exhibits tuneability via control over block composition, molecular weight and cosolvents [1]. To fully understand and exploit the properties of self-assembled structures, the pathways of their formation need to be understood [2]. So far, such a study has been exceedingly difficult if not impossible because of the lack of experimental techniques that are able to capture and resolve the early stage of this rapid process.
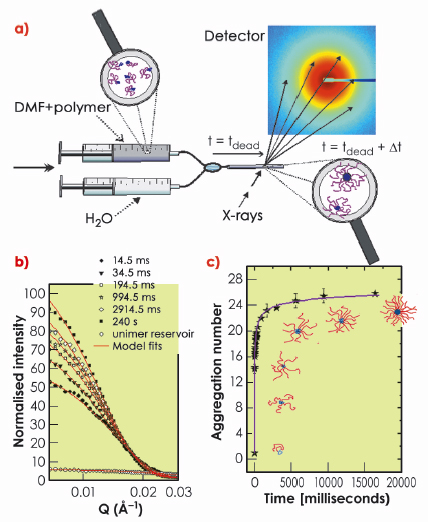
Here we have taken advantage of advances in modern synchrotron radiation instrumentation and have for the first time been able to capture and describe how self-assembly of amphiphilic block copolymers takes place in real time using beamline ID02. An important prerequisite has been to use a well-defined model system consisting of poly(ethylene-alt-propylene)-poly(ethylene oxide) (PEP-PEO) block copolymers in a selective solvent mixture of dimethylformamide and water, both being poor solvents for the PEP block. Block copolymers with large asymmetry in composition (PEP/PEO, 1/20) are molecularly dissolved in pure DMF and micellisation can be induced by simply adding water. This process has been observed experimentally by the setup illustrated in Figure 63, where a stopped flow apparatus [3] is used to realise rapid mixing of the two components (water and polymer in pure DMF solution, respectively) on the millisecond time scale. The reaction itself is monitored directly using fast X-ray shots with some millisecond time resolution that allowed the observation of the birth and growth of the micellar aggregates in time. The evolution of the small-angle scattering for representative times is illustrated in Figure 63b. These curves contain relevant structural characteristics of the micelles, such as sizes, volumes and density profiles. These parameters were extracted by applying detailed core-shell model fits (solid lines). The derived aggregation numbers (number of chains per micelle) are shown in Figure 63c.
 |
|
Fig. 63: Illustration of the experimental setup and the real-time scattering data for the self-assembly process of block copolymers. a) The stopped flow setup consists of two motorised syringes containing the reservoirs with polymer solution (PEP-PEO/DMF) and the solvent (water) respectively. Equal amounts of the solution are mixed and then transferred to the observation capillary within a few milliseconds. b) Normalised absolute scattering data from different times during the kinetic process. c) Extracted aggregation numbers as a function of time. |
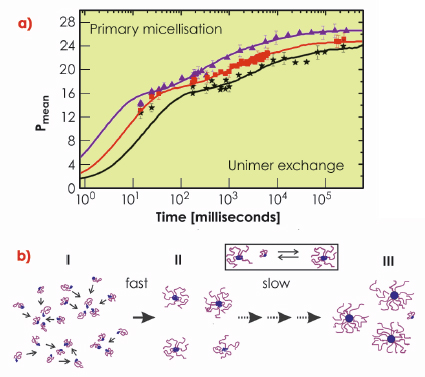
The observed behaviour can be quantitatively reproduced using a kinetic model involving insertion and expulsion of single block polymer chains (unimers) by combining classical nucleation and growth theory [4] with the thermodynamic expression expected for block copolymers. It was assumed that only single molecules (unimers) can be taken up for each cluster at a time. As seen in the simultaneous fits in Figure 64a, the model agrees very well with the experimental data. In the beginning, a very fast initial nucleation, or primary micellisation, that consumes all the unimers can be observed. This leads temporarily almost to a halt of further growth, which is reflected by the plateau like feature visible at intermediate times in Figure 64a. The final stage of the micellisation is governed by unimer exchange following a type of ripening mechanism where small micelles slowly dissolve to provide further unimers while the larger ones continue to grow. This goes on until the micelles approach the shallow minimum of the equilibrium size and the distribution narrows to reflect the thermodynamic equilibrium. This scenario is summarised schematically in Figure 64b.
 |
|
Fig. 64: a) Time evolution of the mean aggregation number of micelles (Pmean) for different concentrations of the block copolymer. Continuous lines represent fits to the nucleation and growth model. b) Schematic representation of the micellisation process involving fast nucleation in which the unimer concentration is depleted followed by slow growth to micelle/unimer equilibrium. |
The excellent agreement between experimental data and this model strongly suggests that the most effective way for micelle formation is simple addition of unimers from a homogeneous solution. This insight gives novel and valuable information of not only the formation and kinetic pathways of these structures but also the stability and lifetime of metastable nano-particles. This knowledge may be utilised for facile predictive design and manipulation of nano-structures, for example in medicine or material science.
References
[1] R. Lund, L. Willner, J. Stellbrink, P. Lindner, and D. Richter, Phys. Rev. Lett. 96, 068302 (2006).
[2] I.A. Nyrkova and A.N. Semenov, Macromol. Theory and Sim. 14, 569 (2005).
[3] P. Panine, S. Finet, T. Weiss, and T. Narayanan, Adv. Colloid Interf. Sci. 127, 9 (2006).
[4] J.C. Neu, J.A. Cañizo and L.L. Bonilla, Phys. Rev. E 66, 061406 (2002).
Principal publication and authors
R. Lund (a), L. Willner (b), M. Monkenbusch (b), P. Panine (c), T. Narayanan (c), J. Colmenero (a) and D. Richter (b), Phys. Rev Lett. 102, 188301 (2009).
(a) Donostia International Physics Center/University of the Basque Country, San Seabastian (Spain)
(b) Institute of Solid State Research (IFF), Forschungszentrum Jülich (Germany)
(c) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.