- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2009
- Structure of materials
- Structure and reactivity of Rh oxides
Structure and reactivity of Rh oxides
More than 80% of all chemical products are produced using catalysts, which facilitate a specific reaction without being consumed. In most cases the catalyst is in a different phase to the reactants, e.g. a solid catalyst and gaseous reactants, a situation referred to as heterogeneous catalysis. Industrial catalysts are complicated materials systems consisting of oxide supports with dispersed metal nanoparticles of the active catalyst as well as a wide range of additives to promote or poison specific reactions. Owing to their complexity, information on the functioning of the catalyst at the atomic scale is at best limited, and consequently most catalyst development is done via an empirical approach.
Much effort has recently been directed towards replicating the conditions under which most industrial catalysts work. In such in situ studies, the structure is analysed during the reaction under realistic pressures. We have used the batch reactor surface X-ray diffraction (SXRD) setup at beamline ID03 to follow the oxidation of Rh – one of the active elements in a car’s exhaust-gas catalytic converter – and investigated how the catalytic activity varies with the oxidation state. Prior to the formation of a thick bulk oxide, an ultrathin surface oxide is formed, exhibiting a trilayer structure with two hexagonal oxygen layers separated by a single Rh layer. This surface oxide does not grow under UHV compatible conditions, but is found on all surface orientations at intermediate oxygen pressures.
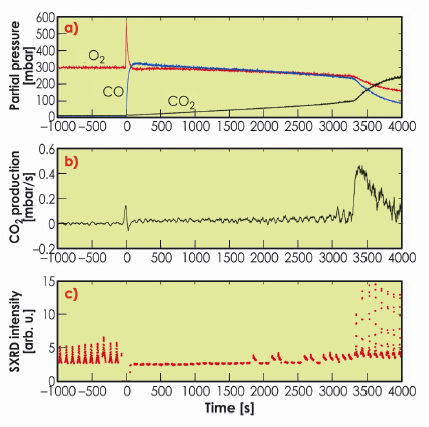
Figure 38 shows the results from the oxidation of CO over Rh(111). At t = –1000 s, 300 mbar of O2 and close to no CO and CO2 are present in the batch reactor. At this time, SXRD shows the presence of the surface oxide. At time t = 0, 330 mbar CO is added, and the CO2 production starts. Here, no surface oxide can be detected. As one O2 molecule can oxidise two CO molecules, the O2/CO partial pressure ratio increases as the reaction proceeds, creating a more oxidising environment. At t ~ 3300 s, we notice a significant increase in the reaction rate, which coincides with the return of the surface oxide. We have found very similar results, where the Rh surface oxide shows a very high CO oxidation reactivity, for Rh(100) as well as Pt25Rh75(100) samples. The bulk oxide, however, is found to be inactive.
 |
|
Fig. 38: Activity and surface structure of Rh(111) during CO oxidation. a) the partial pressure of O2, CO and CO2, b) the differentiated CO2 signal, i.e. the CO2 production rate, c) consecutive SXRD scans revealing the presence (or absence) of the surface oxide. |
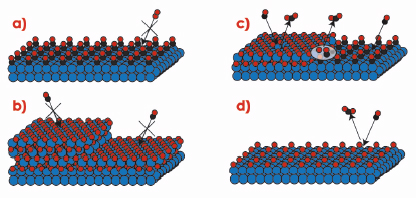
The difference in reactivity between the oxidised and metallic surfaces is striking, and indicates that the surface oxide plays a crucial role in the high activity of Rh-based CO oxidation catalysts. Under UHV conditions, however, CO has a very low adsorption energy on the surface oxide, which would imply that the surface oxide rather poisons the catalyst. In order to find an explanation for the behaviour at elevated pressures, Figure 39 shows a number of different phases, in which the catalyst surface can be found. In Figure 39a the surface is in a metallic phase covered by adsorbed CO molecules. Since the O2 adsorption is dissociative it needs a larger free surface area than can be found between the CO molecules, which makes this phase inactive. Similarly, the oxide surfaces shown in Figure 39b are fully covered by O atoms, which does not allow for CO adsorption, this also resulting in inactive surfaces. To make the surface oxide active, either defects or co-existing metallic areas, where the CO can adsorb, are needed (Figure 39c). Alternatively a surface with adsorbed O atoms, as shown in Figure 39d, will still allow CO adsorption (which is not dissociative) and be active. According to density functional theory calculations, however, the reaction barriers involved in forming CO2 using chemisorbed O is significantly higher than those for using O from the edge of the surface oxide [1]. Hence the reactivity is expected to be lower in case (d) when compared to (c).
 |
|
Fig. 39: Different phases in which the Rh catalyst can be found in an atmosphere of O2 and CO. |
In summary, our study indicates that structures that only form under realistic operating conditions may be of major importance for a fundamental understanding of catalysis.
Reference
[1] R. Westerström, J.G. Wang, M.D. Ackermann, J. Gustafson, A. Resta, A. Mikkelsen, J.N. Andersen, E. Lundgren, O. Balmes, X. Torrelles, J.W.M. Frenken and B. Hammer, J. Phys. Condens. Matter 20, 184018 (2008).
Principal publications and authors
J. Gustafson (a,b), R. Westerström (a), A. Resta (a,c), A. Mikkelsen (a), J.N. Andersen (a), O. Balmes (c), X. Torrelles (d), M. Schmid (e), P. Varga (e), B. Hammer (f), G. Kresse (g), C.J. Baddeley (b) and E. Lundgren (a), Cat. Today 145, 227-235 (2009);
J. Gustafson (a,b), R. Westerström (a), A. Mikkelsen (a), X. Torrelles (d), O. Balmes (c), N. Bovet (h), J.N. Andersen (a), C.J. Baddeley (b), and E. Lundgren (a), Phys. Rev. B 78, 045423 (2008).
(a) Department of Synchrotron Radiation Research, Lund University (Sweden)
(b) EaStCHEM School of Chemistry, University of St. Andrews (UK)
(c) ESRF
(d) Instituto de Ciencia de Materiales de Barcelona (Spain)
(e) Institut für Algemeine Physik, Technische Universität Wien (Austria)
(f) Interdisiplinary Nanoscience Center (iNANO) and Department of Physics and Astronomy, University of Aarhus (Denmark)
(g) Institut für Materialphysik, Universität Wien (Austria)
(h) MAX-lab, Lund University (Sweden)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.