- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2009
- Structural biology
- Complement activation and regulation based on crystal structures of (C3bBb-SCIN)2 and C3b-factor H(1-4)
Complement activation and regulation based on crystal structures of (C3bBb-SCIN)2 and C3b-factor H(1-4)
The complement cascade is an ancient defence mechanism of the mammalian immune system that consists of more than 30 proteins circulating in blood and fluids surrounding tissues or present on cell surfaces. Activation of the complement system generates potent chemoattractants and markers for immune clearance that covalently attach to cell surfaces. Amplification of this process by the alternative pathway is mainly determined by the formation of short-lived protease complexes, known as C3 convertases (half-life time of ~90 seconds). C3 convertase cleaves the central complement component C3 into its active form C3b, which exposes its thioester bond through which it covalently attaches to nearby surfaces. This process must be tightly controlled by complement regulators in order to prevent non-specific attack of host cells. These regulators stop C3b production either by accelerating the decay of the C3 convertases or by serving as a cofactor for protease factor I (FI) in degrading existing C3b (thereby preventing formation of new convertases). Factor H (FH) is a highly abundant, soluble complement regulator, which can either function in the fluid phase or on the host cell surface by recognition of host specific components, such as glycosaminoglycans.
 |
|
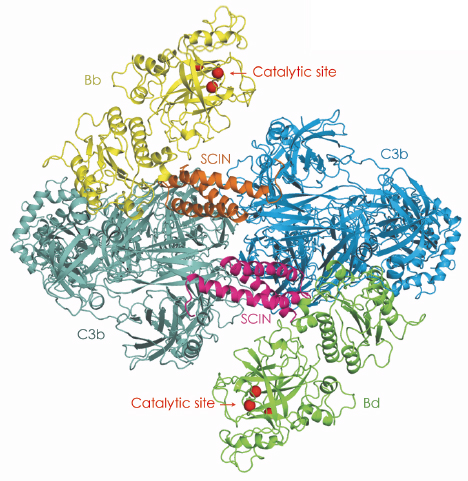
Fig. 104: Crystal structure of the labile C3 convertase (C3bBb) stabilised by the staphylococcal complement inhibitor SCIN. The observed dimeric arrangement suggests that substrate specificity and catalytic activity is determined by dimerisation of the substrate C3 and ligand C3b. |
The unstable C3 convertase complex (of the alternative pathway) is formed by the 12-domain ligand C3b and the 2-domain protease fragment Bb. An immune evasion protein SCIN secreted by Staphylococcus aureus was used to stabilise the C3 convertase. The structure of the resulting complex (C3bBb-SCIN)2, with MW ~500 kDa, was determined at 3.9 Å resolution using diffraction data collected on beamline ID14-4. In the dimer arrangement seen in the crystal, the two convertase complexes are arranged with C2 symmetry (Figure 104). In each monomer, the protease fragment Bb is attached to the C-terminal Asn 1641 of C3b. This residue chelates the Mg2+ bound in the metal ion-dependent adhesion site (MIDAS) in Bb. SCIN stabilises dimer formation of the C3 convertase complex by bridging the opposing C3b molecules. The dimeric arrangement yields unprecedented structural insights into the unique substrate specificity and enzymatic activity of this protease complex. Substitution of one C3b by the substrate C3 places the scissile bond of C3 in front of the active site of the opposing C3bBb complex. This hypothetical enzyme:substrate model explains the activity of three inhibitors that block the substrate binding to C3 convertases. Thus, specificity and activity of the C3 convertase is likely to be determined by dimerisation of the substrate C3 with the cleavage product C3b that serves as a ligand in the C3bBb protease.
 |
|
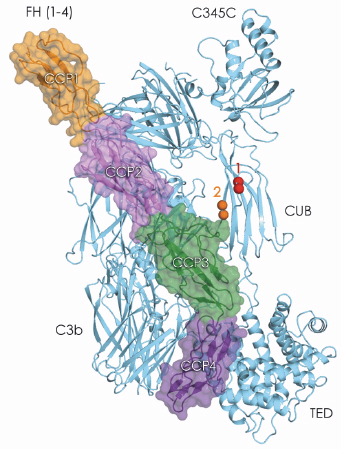
Fig. 105: Crystal structure of C3b in complex with FH domains 1-4. FH domains 1-2 are likely to be directly responsible for displacing the protease fragment Bb from the C3bBb complex. In addition, C3bFH provide a binding place for the protease FI, putatively involving CCP1-3 of FH and domains C345C and CUB of C3b. FI cleaves the chain of the CUB domain of C3b twice (scissile bonds are indicated in spheres) to produce inactive iC3b. |
To gain insight into complement regulation, we solved the crystal structure of C3b in complex with the first four ‘complement-control protein’ (CCP) domains of FH (FH(1-4)) to 2.7 Å resolution, using data collected at ID14-4. The structure reveals an extensive and discontinuous interface between FH(1-4) and C3b that stretches over a distance of 100 Å and buries an area of 4,500 Å2 (Figure 105). Comparison of C3b-FH(1-4) and C3bBb indicates a functional role for domains CCP1-2 of FH in displacing Bb from C3b, which underpins the ‘decay acceleration activity’. Moreover, FH(1-4) binds adjacent to the CUB domain of C3b that contains two cleavage sites for the protease FI. This arrangement and biochemical data suggest that in ‘cofactor activity’ FH domains CCP1-3 and possibly C345C and CUB of C3b form the binding site for FI. Additional contacts between CCP4 and the thioester domain may stabilise the overall arrangement while the protease makes consecutive cuts in the chain of the bridging CUB domain.
Principal publication and authors
J. Wu (a), S.H.M. Rooijakkers (b), M. Ruyken (b), R. van Domselaar (b), K.L. Planken (c), A. Tzekou (d), D. Ricklin (d), J.D. Lambris (d), B.J.C. Janssen (a), J.A.G. van Strijp (b) and P. Gros (a), Nature Immunology 10, 721-727 (2009); J. Wu (a), Y.-Q. Wu (d), D. Ricklin (d), B.J.C. Janssen (a), J.D. Lambris (d) and P. Gros (a), Nature Immunology 10, 728-733 (2009).
(a) Crystal and Structural Chemistry, Bijvoet Center, Utrecht University (The Netherlands)
(b) Medical Microbiology, University Medical Center Utrecht (The Netherlands)
(c) Van ‘t Hoff Laboratory for Physical and Colloid Chemistry, Utrecht University (The Netherlands)
(d) Department of Pathology & Laboratory Medicine, University of Pennsylvania (USA)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.